

A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition
- PMID: 27525524
- PMCID: PMC5706766
- DOI: 10.1038/nm.4170
A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition
Abstract
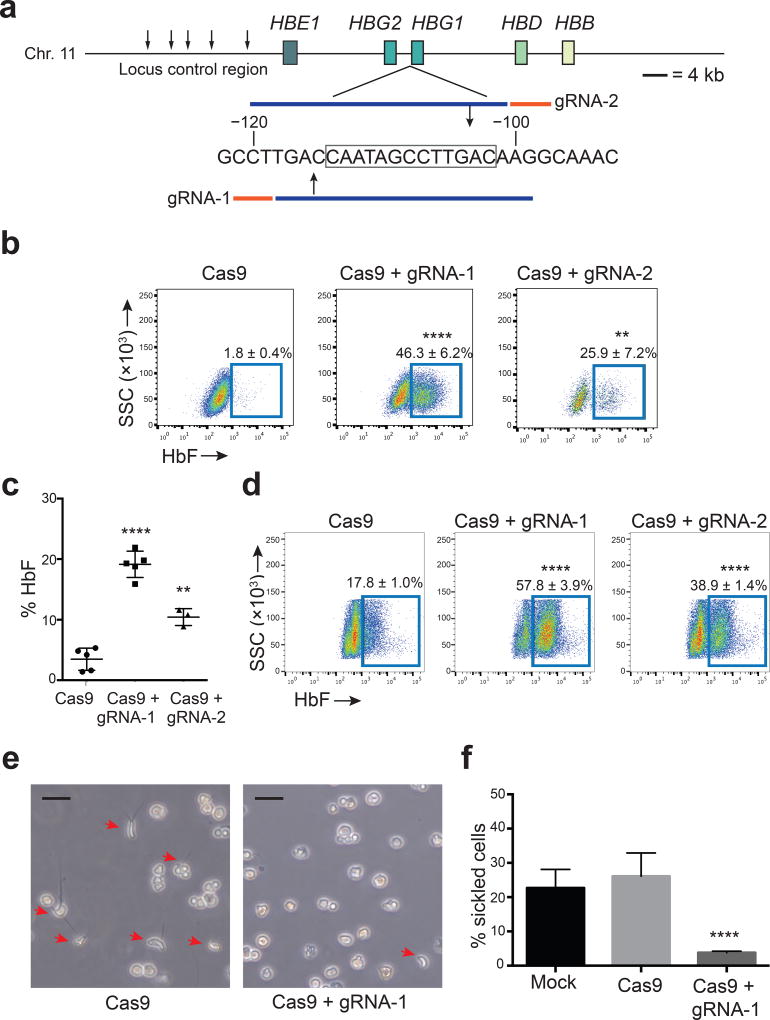
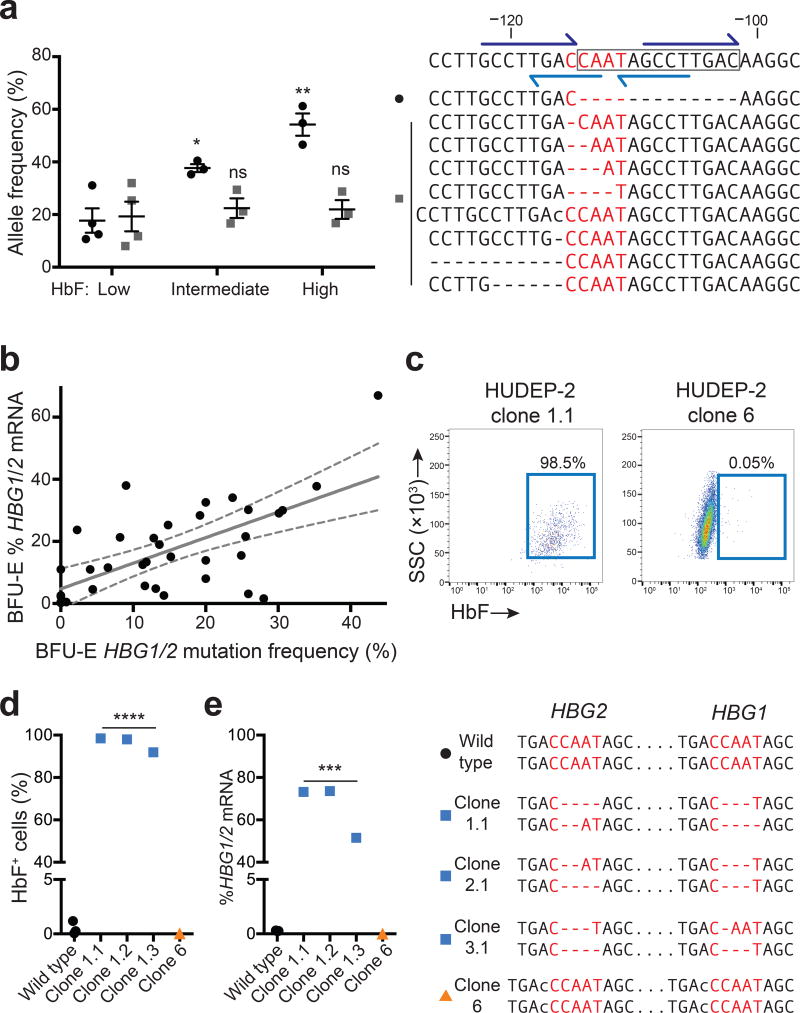
Disorders resulting from mutations in the hemoglobin subunit beta gene (HBB; which encodes β-globin), mainly sickle cell disease (SCD) and β-thalassemia, become symptomatic postnatally as fetal γ-globin expression from two paralogous genes, hemoglobin subunit gamma 1 (HBG1) and HBG2, decreases and adult β-globin expression increases, thereby shifting red blood cell (RBC) hemoglobin from the fetal (referred to as HbF or α2γ2) to adult (referred to as HbA or α2β2) form. These disorders are alleviated when postnatal expression of fetal γ-globin is maintained. For example, in hereditary persistence of fetal hemoglobin (HPFH), a benign genetic condition, mutations attenuate γ-globin-to-β-globin switching, causing high-level HbF expression throughout life. Co-inheritance of HPFH with β-thalassemia- or SCD-associated gene mutations alleviates their clinical manifestations. Here we performed CRISPR-Cas9-mediated genome editing of human blood progenitors to mutate a 13-nt sequence that is present in the promoters of the HBG1 and HBG2 genes, thereby recapitulating a naturally occurring HPFH-associated mutation. Edited progenitors produced RBCs with increased HbF levels that were sufficient to inhibit the pathological hypoxia-induced RBC morphology found in SCD. Our findings identify a potential DNA target for genome-editing-mediated therapy of β-hemoglobinopathies.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Forget BG. Molecular basis of hereditary persistence of fetal hemoglobin. Ann. N. Y. Acad. Sci. 1998;850:38–44. - PubMed
-
- Wienert B, et al. Editing the genome to introduce a beneficial naturally occurring mutation associated with increased fetal globin. Nat Commun. 2015;6:7085. - PubMed
-
- Liberati C, et al. Cooperation and competition between the binding of COUP-TFII and NF-Y on human epsilon- and gamma-globin gene promoters. J Biol Chem. 2001;276:41700–41709. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
