

Comprehensive Screening of Eight Known Causative Genes in Congenital Hypothyroidism With Gland-in-Situ
- PMID: 27525530
- PMCID: PMC5155683
- DOI: 10.1210/jc.2016-1879
Comprehensive Screening of Eight Known Causative Genes in Congenital Hypothyroidism With Gland-in-Situ
Abstract
Context: Lower TSH screening cutoffs have doubled the ascertainment of congenital hypothyroidism (CH), particularly cases with a eutopically located gland-in-situ (GIS). Although mutations in known dyshormonogenesis genes or TSHR underlie some cases of CH with GIS, systematic screening of these eight genes has not previously been undertaken.
Objective: Our objective was to evaluate the contribution and molecular spectrum of mutations in eight known causative genes (TG, TPO, DUOX2, DUOXA2, SLC5A5, SLC26A4, IYD, and TSHR) in CH cases with GIS. Patients, Design, and Setting: We screened 49 CH cases with GIS from 34 ethnically diverse families, using next-generation sequencing. Pathogenicity of novel mutations was assessed in silico.
Patients, design, and setting: We screened 49 CH cases with GIS from 34 ethnically diverse families, using next-generation sequencing. Pathogenicity of novel mutations was assessed in silico.
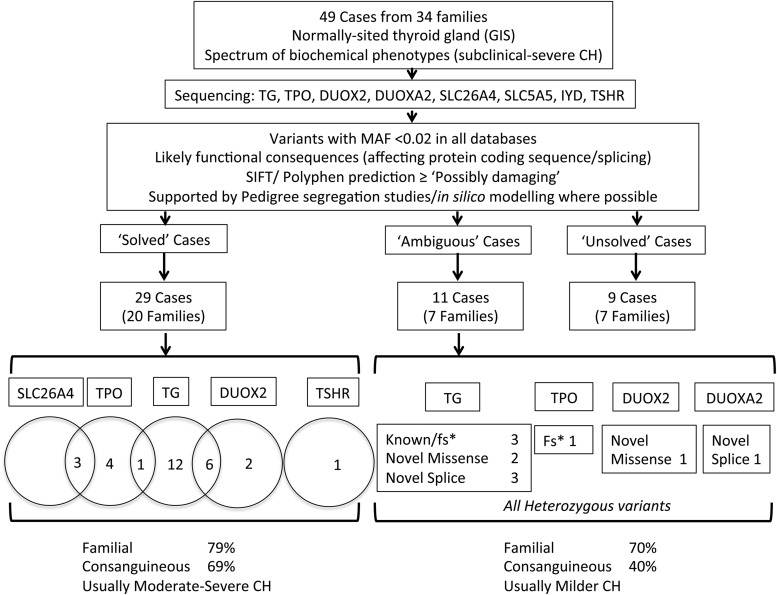
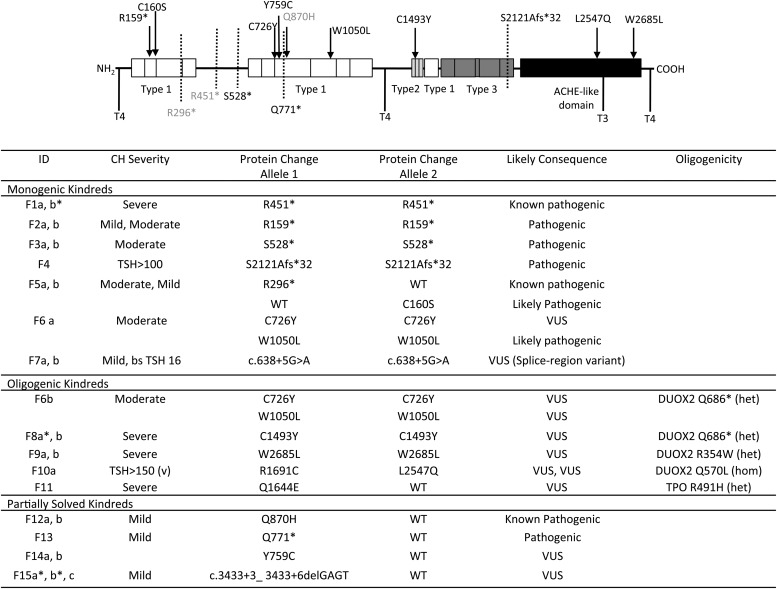
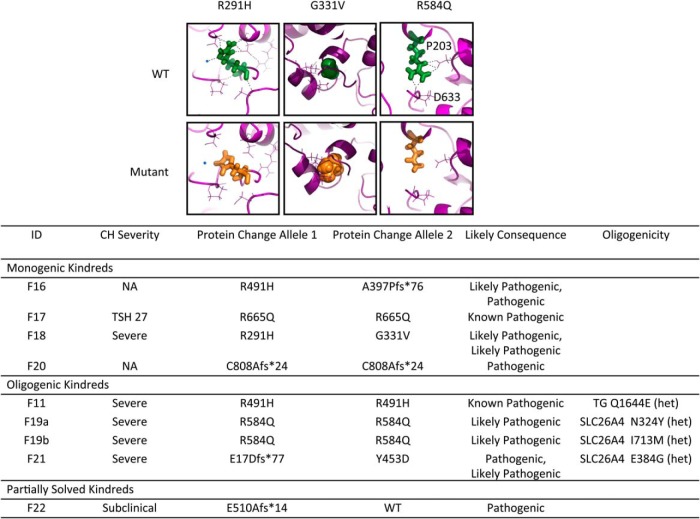
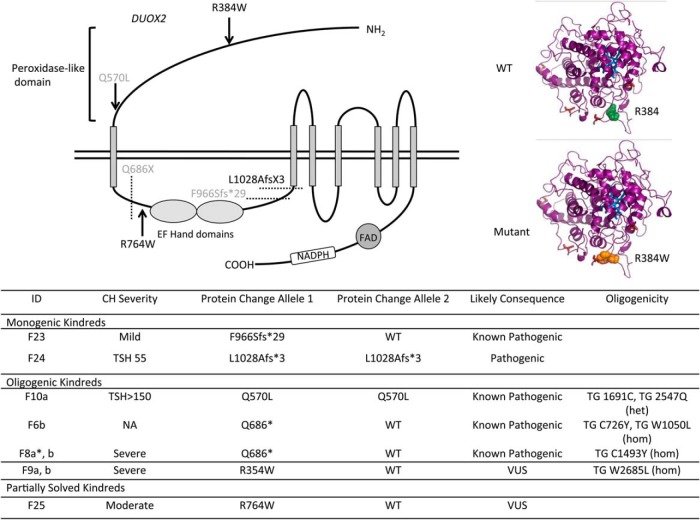
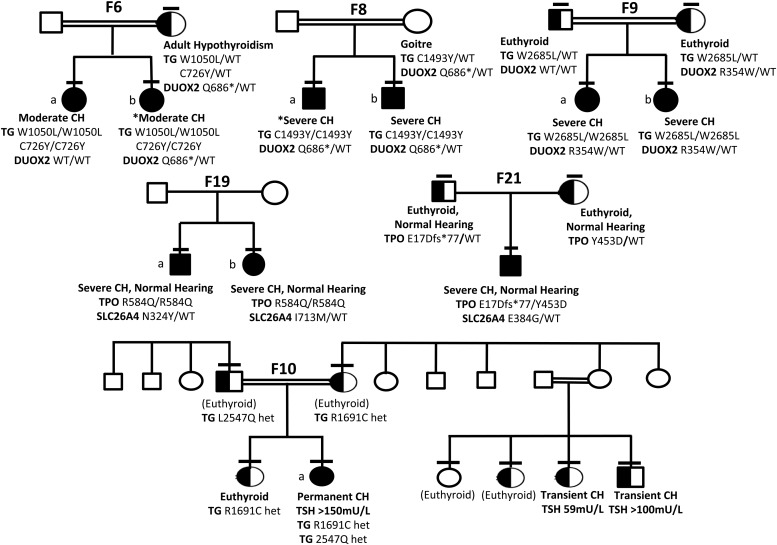
Results: Twenty-nine cases harbored likely disease-causing mutations. Monogenic defects (19 cases) most commonly involved TG (12), TPO (four), DUOX2 (two), and TSHR (one). Ten cases harbored triallelic (digenic) mutations: TG and TPO (one); SLC26A4 and TPO (three), and DUOX2 and TG (six cases). Novel variants overall included 15 TG, six TPO, and three DUOX2 mutations. Genetic basis was not ascertained in 20 patients, including 14 familial cases.
Conclusions: The etiology of CH with GIS remains elusive, with only 59% attributable to mutations in TSHR or known dyshormonogenesis-associated genes in a cohort enriched for familial cases. Biallelic TG or TPO mutations most commonly underlie severe CH. Triallelic defects are frequent, mandating future segregation studies in larger kindreds to assess their contribution to variable phenotype. A high proportion (∼41%) of unsolved or ambiguous cases suggests novel genetic etiologies that remain to be elucidated.
Figures
References
-
- Szinnai G. 2014 clinical genetics of congenital hypothyroidism. Paediatric Thyroidology. Endocr Dev. Basel, Karger, 2014;26:60–78. - PubMed
-
- Corbetta C, Weber G, Cortinovis F, et al. A 7-year experience with low blood TSH cutoff levels for neonatal screening reveals an unsuspected frequency of congenital hypothyroidism (CH). Clin Endocrinol (Oxf). 2009;71:739–745. - PubMed
-
- Harris KB, Pass KA. Increase in congenital hypothyroidism in New York State and in the United States. Mol Genet Metab. 2007;91:268–277. - PubMed
-
- Rabbiosi S, Vigone MC, Cortinovis F, et al. Congenital hypothyroidism with eutopic thyroid gland: analysis of clinical and biochemical features at diagnosis and after re-evaluation. J Clin Endocrinol Metab. 2013;98:1395–1402. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
