

Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution
- PMID: 27526324
- PMCID: PMC5042844
- DOI: 10.1038/ng.3646
Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution
Abstract
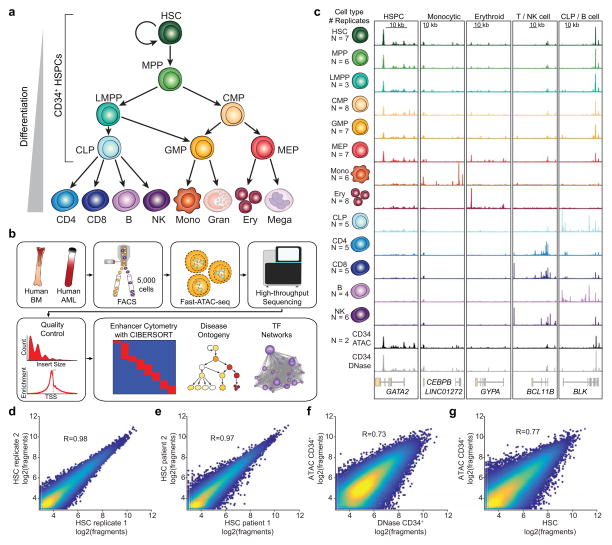
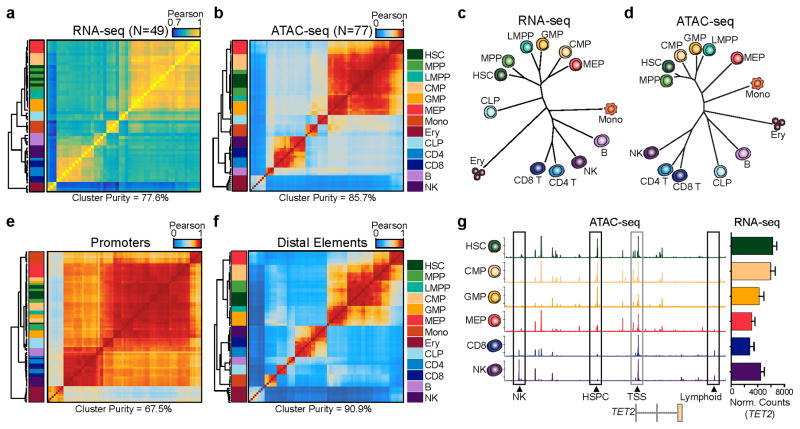
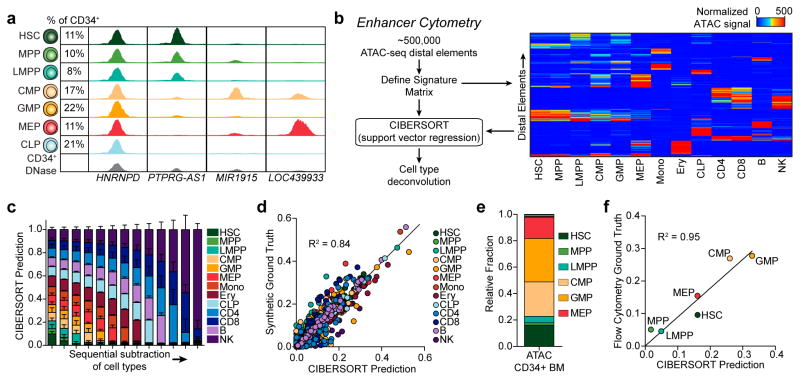
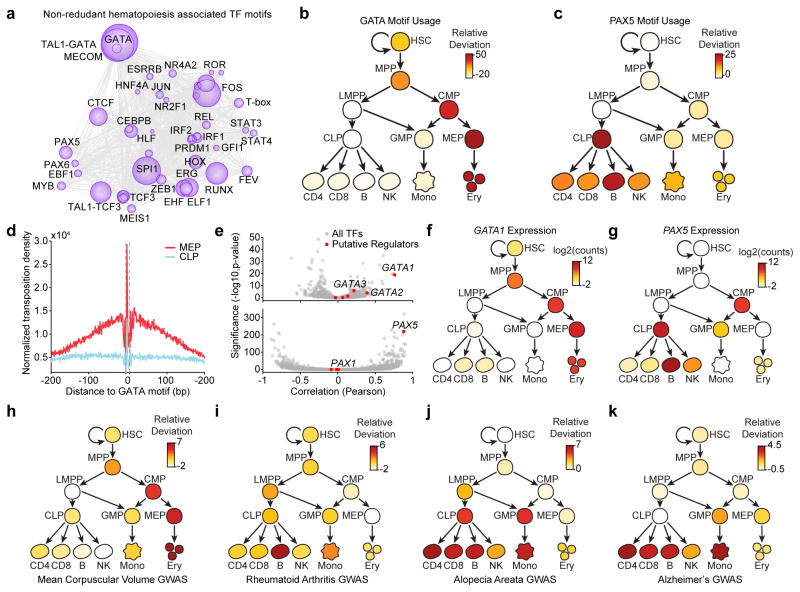
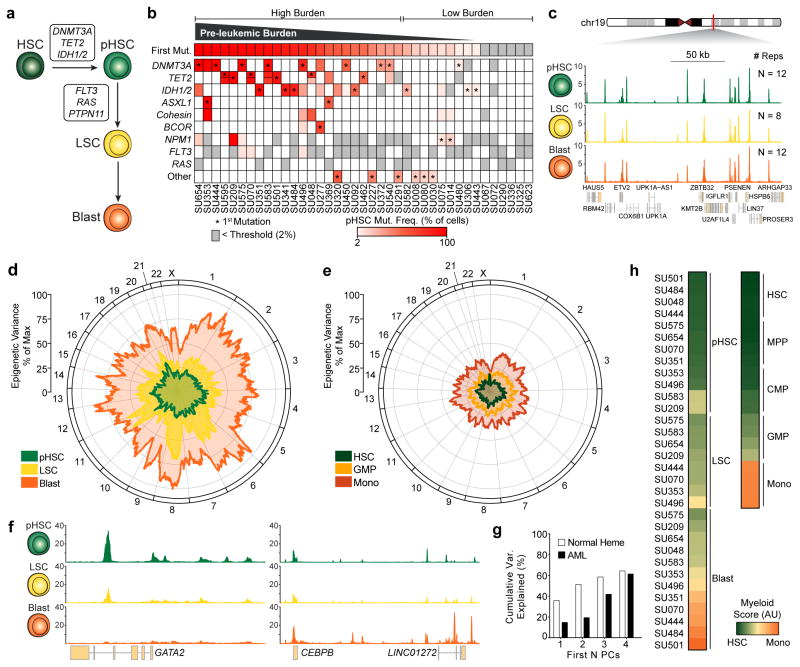
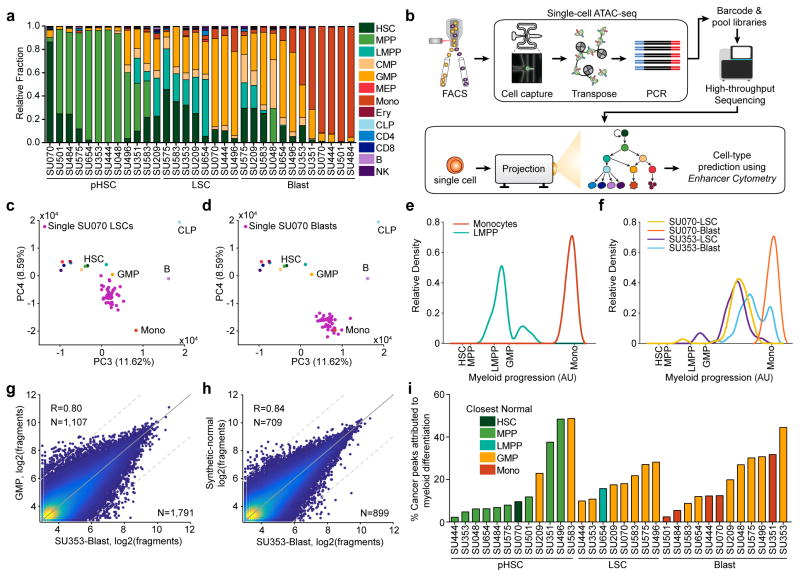
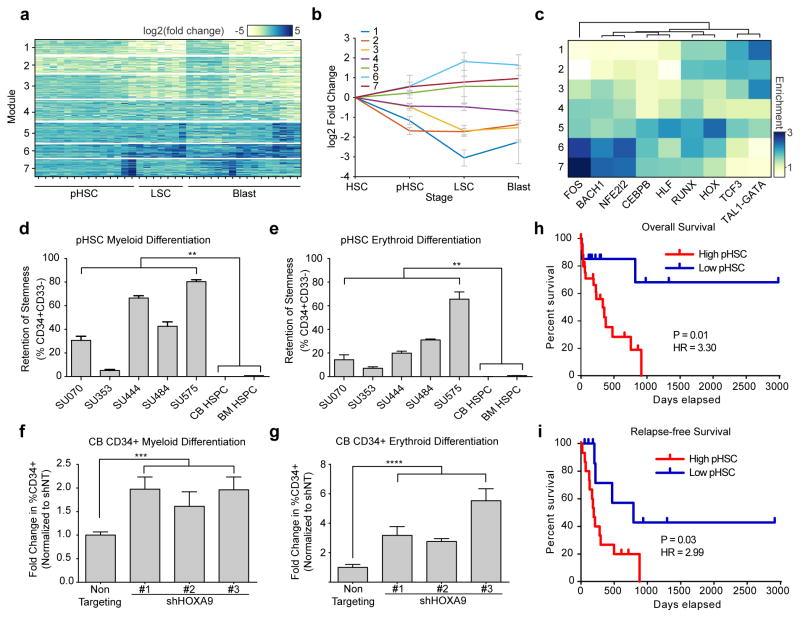
We define the chromatin accessibility and transcriptional landscapes in 13 human primary blood cell types that span the hematopoietic hierarchy. Exploiting the finding that the enhancer landscape better reflects cell identity than mRNA levels, we enable 'enhancer cytometry' for enumeration of pure cell types from complex populations. We identify regulators governing hematopoietic differentiation and further show the lineage ontogeny of genetic elements linked to diverse human diseases. In acute myeloid leukemia (AML), chromatin accessibility uncovers unique regulatory evolution in cancer cells with a progressively increasing mutation burden. Single AML cells exhibit distinctive mixed regulome profiles corresponding to disparate developmental stages. A method to account for this regulatory heterogeneity identified cancer-specific deviations and implicated HOX factors as key regulators of preleukemic hematopoietic stem cell characteristics. Thus, regulome dynamics can provide diverse insights into hematopoietic development and disease.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Quesenberry PJ, Colvin GA. Williams Hematology. McGraw-Hill; 2005. Hematopoietic Stem Cells, Progenitor Cells, and Cytokines.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
