

Differences and Similarities in TRAIL- and Tumor Necrosis Factor-Mediated Necroptotic Signaling in Cancer Cells
- PMID: 27528614
- PMCID: PMC5038143
- DOI: 10.1128/MCB.00941-15
Differences and Similarities in TRAIL- and Tumor Necrosis Factor-Mediated Necroptotic Signaling in Cancer Cells
Abstract
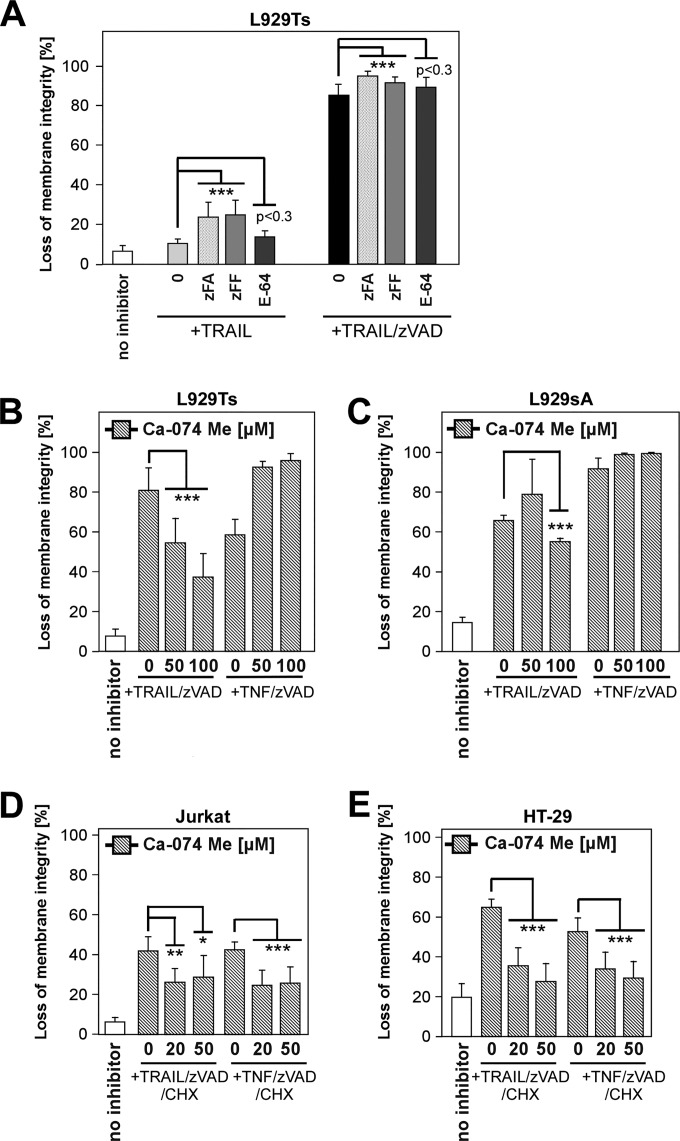
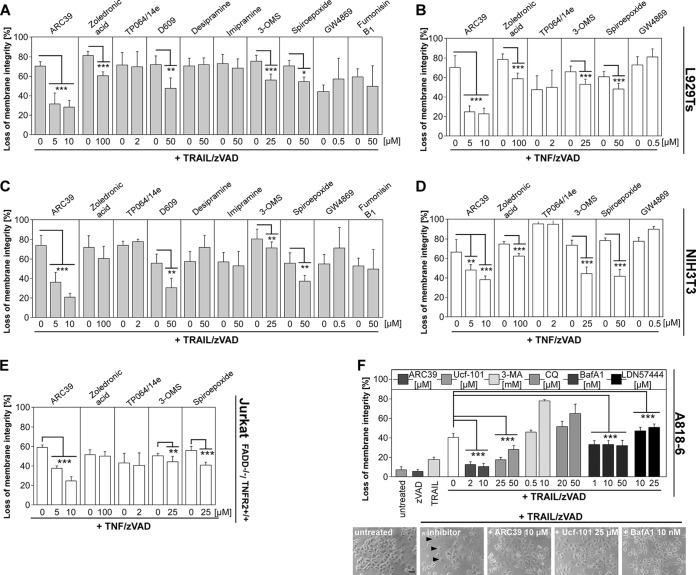
Recently, a type of regulated necrosis (RN) called necroptosis was identified to be involved in many pathophysiological processes and emerged as an alternative method to eliminate cancer cells. However, only a few studies have elucidated components of TRAIL-mediated necroptosis useful for anticancer therapy. Therefore, we have compared this type of cell death to tumor necrosis factor (TNF)-mediated necroptosis and found similar signaling through acid and neutral sphingomyelinases, the mitochondrial serine protease HtrA2/Omi, Atg5, and vacuolar H(+)-ATPase. Notably, executive mechanisms of both TRAIL- and TNF-mediated necroptosis are independent of poly(ADP-ribose) polymerase 1 (PARP-1), and depletion of p38α increases the levels of both types of cell death. Moreover, we found differences in signaling between TNF- and TRAIL-mediated necroptosis, e.g., a lack of involvement of ubiquitin carboxyl hydrolase L1 (UCH-L1) and Atg16L1 in executive mechanisms of TRAIL-mediated necroptosis. Furthermore, we discovered indications of an altered involvement of mitochondrial components, since overexpression of the mitochondrial protein Bcl-2 protected Jurkat cells from TRAIL- and TNF-mediated necroptosis, and overexpression of Bcl-XL diminished only TRAIL-induced necroptosis in Colo357 cells. Furthermore, TRAIL does not require receptor internalization and endosome-lysosome acidification to mediate necroptosis. Taken together, pathways described for TRAIL-mediated necroptosis and differences from those for TNF-mediated necroptosis might be unique targets to increase or modify necroptotic signaling and eliminate tumor cells more specifically in future anticancer approaches.
Copyright © 2016, American Society for Microbiology. All Rights Reserved.
Figures








Similar articles
-
The proteases HtrA2/Omi and UCH-L1 regulate TNF-induced necroptosis.Cell Commun Signal. 2013 Oct 3;11:76. doi: 10.1186/1478-811X-11-76. Cell Commun Signal. 2013. PMID: 24090154 Free PMC article.
-
Homoharringtonine, a clinically approved anti-leukemia drug, sensitizes tumor cells for TRAIL-induced necroptosis.Cell Commun Signal. 2015 Apr 29;13:25. doi: 10.1186/s12964-015-0103-0. Cell Commun Signal. 2015. PMID: 25925126 Free PMC article.
-
Casein kinase I attenuates tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by regulating the recruitment of fas-associated death domain and procaspase-8 to the death-inducing signaling complex.Cancer Res. 2004 Nov 1;64(21):8036-44. doi: 10.1158/0008-5472.CAN-04-0762. Cancer Res. 2004. PMID: 15520213
-
Cancer and necroptosis: friend or foe?Cell Mol Life Sci. 2016 Jun;73(11-12):2183-93. doi: 10.1007/s00018-016-2193-2. Epub 2016 Apr 5. Cell Mol Life Sci. 2016. PMID: 27048810 Free PMC article. Review.
-
Paving TRAIL's Path with Ubiquitin.Trends Biochem Sci. 2018 Jan;43(1):44-60. doi: 10.1016/j.tibs.2017.11.002. Epub 2017 Nov 28. Trends Biochem Sci. 2018. PMID: 29195774 Review.
Cited by
-
Protease-independent control of parthanatos by HtrA2/Omi.Cell Mol Life Sci. 2023 Aug 18;80(9):258. doi: 10.1007/s00018-023-04904-7. Cell Mol Life Sci. 2023. PMID: 37594630 Free PMC article.
-
Regulation of Death Receptor Signaling by S-Palmitoylation and Detergent-Resistant Membrane Micro Domains-Greasing the Gears of Extrinsic Cell Death Induction, Survival, and Inflammation.Cancers (Basel). 2021 May 21;13(11):2513. doi: 10.3390/cancers13112513. Cancers (Basel). 2021. PMID: 34063813 Free PMC article. Review.
-
Contribution of cell death signaling to blood vessel formation.Cell Mol Life Sci. 2021 Apr;78(7):3247-3264. doi: 10.1007/s00018-020-03738-x. Epub 2021 Mar 30. Cell Mol Life Sci. 2021. PMID: 33783563 Free PMC article. Review.
-
Assessing Autophagy in Archived Tissue or How to Capture Autophagic Flux from a Tissue Snapshot.Biology (Basel). 2020 Mar 21;9(3):59. doi: 10.3390/biology9030059. Biology (Basel). 2020. PMID: 32245178 Free PMC article.
-
Proteasome inhibition blocks necroptosis by attenuating death complex aggregation.Cell Death Dis. 2018 Mar 1;9(3):346. doi: 10.1038/s41419-018-0371-x. Cell Death Dis. 2018. PMID: 29497034 Free PMC article.
References
-
- Jin Z, El-Deiry WS. 2005. Overview of cell death signaling pathways. Cancer Biol Ther 4:139–163. - PubMed
-
- Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, Hengartner M, Knight RA, Kumar S, Lipton SA, Malorni W, Nunez G, Peter ME, Tschopp J, Yuan J, Piacentini M, Zhivotovsky B, Melino G, Nomenclature Committee on Cell Death 2009. 2009. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 16:3–11. doi:10.1038/cdd.2008.150. - DOI - PMC - PubMed
-
- Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, Gottlieb E, Green DR, Hengartner MO, Kepp O, Knight RA, Kumar S, Lipton SA, Lu X, Madeo F, Malorni W, Mehlen P, Nunez G, Peter ME, Piacentini M, Rubinsztein DC, Shi Y, Simon HU, Vandenabeele P, White E, Yuan J, Zhivotovsky B, Melino G, Kroemer G. 2012. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 19:107–120. doi:10.1038/cdd.2011.96. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous