

Modulating mitochondrial quality in disease transmission: towards enabling mitochondrial DNA disease carriers to have healthy children
- PMID: 27528757
- PMCID: PMC4984448
- DOI: 10.1042/BST20160095
Modulating mitochondrial quality in disease transmission: towards enabling mitochondrial DNA disease carriers to have healthy children
Abstract
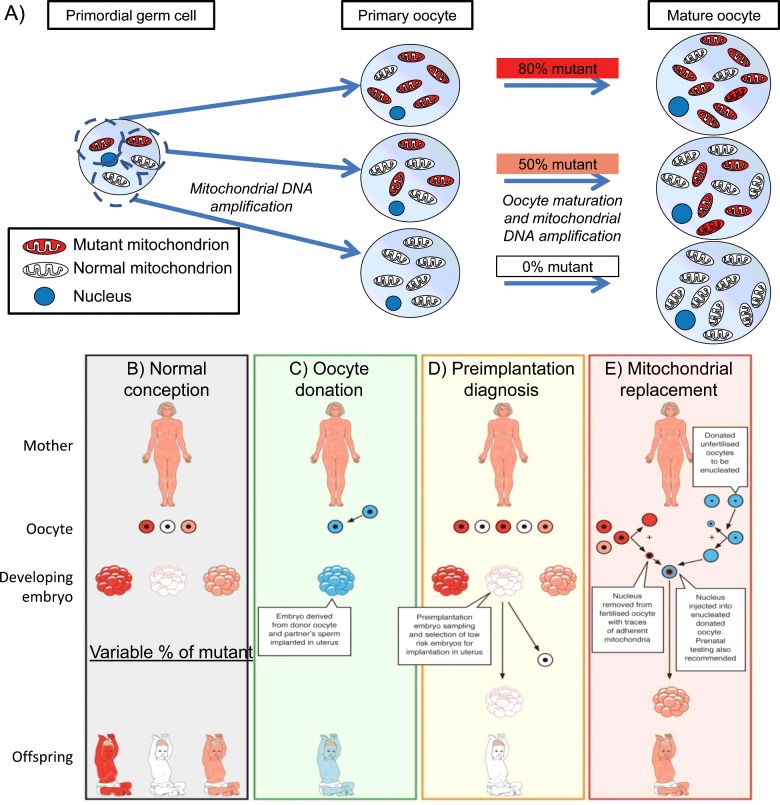
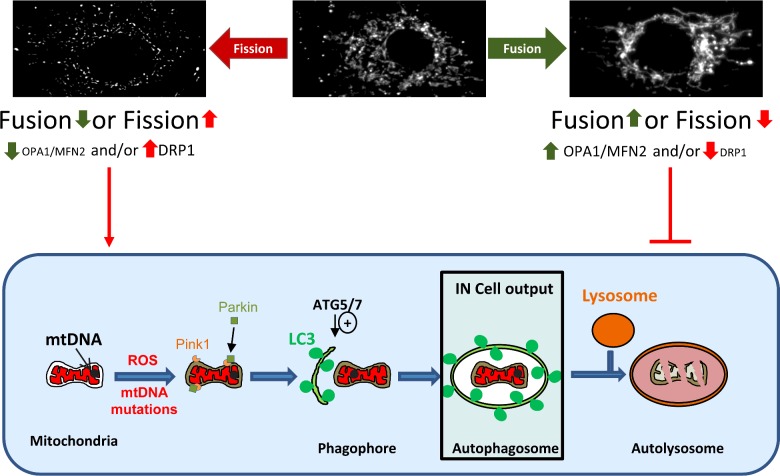
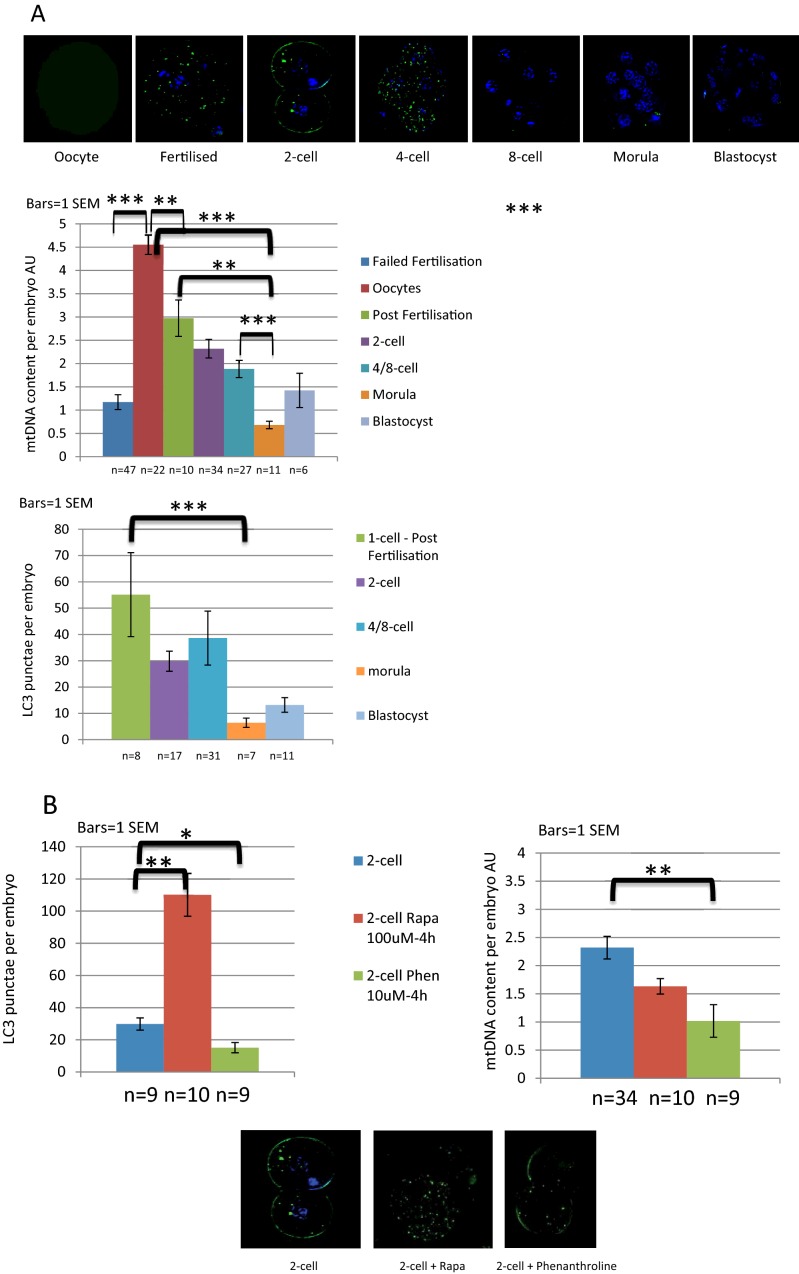
One in 400 people has a maternally inherited mutation in mtDNA potentially causing incurable disease. In so-called heteroplasmic disease, mutant and normal mtDNA co-exist in the cells of carrier women. Disease severity depends on the proportion of inherited abnormal mtDNA molecules. Families who have had a child die of severe, maternally inherited mtDNA disease need reliable information on the risk of recurrence in future pregnancies. However, prenatal diagnosis and even estimates of risk are fraught with uncertainty because of the complex and stochastic dynamics of heteroplasmy. These complications include an mtDNA bottleneck, whereby hard-to-predict fluctuations in the proportions of mutant and normal mtDNA may arise between generations. In 'mitochondrial replacement therapy' (MRT), damaged mitochondria are replaced with healthy ones in early human development, using nuclear transfer. We are developing non-invasive alternatives, notably activating autophagy, a cellular quality control mechanism, in which damaged cellular components are engulfed by autophagosomes. This approach could be used in combination with MRT or with the regular management, pre-implantation genetic diagnosis (PGD). Mathematical theory, supported by recent experiments, suggests that this strategy may be fruitful in controlling heteroplasmy. Using mice that are transgenic for fluorescent LC3 (the hallmark of autophagy) we quantified autophagosomes in cleavage stage embryos. We confirmed that the autophagosome count peaks in four-cell embryos and this correlates with a drop in the mtDNA content of the whole embryo. This suggests removal by mitophagy (mitochondria-specific autophagy). We suggest that modulating heteroplasmy by activating mitophagy may be a useful complement to mitochondrial replacement therapy.
Keywords: mitochondrial replacement therapy; mitophagy; mtDNA bottleneck.
© 2016 The Author(s).
Figures
References
-
- Steffann J., Frydman N., Gigarel N., Burlet P., Ray P.F., Fanchin R., Feyereisen E., Kerbrat V., Tachdjian G., Bonnefont J.P., et al. Analysis of mtDNA variant segregation during early human embryonic development: a tool for successful NARP preimplantation diagnosis. J. Med. Genet. 2006;43:244–247. doi: 10.1136/jmg.2005.032326. - DOI - PMC - PubMed
-
- Bouchet C., Steffann J., Corcos J., Monnot S., Paquis V., Rotig A., Lebon S., Levy P., Royer G., Giurgea I., et al. Prenatal diagnosis of MELAS syndrome: contribution to understanding mitochondrial DNA segregation during human embryo fetal development. J. Med. Genet. 2006;43:788–792. doi: 10.1136/jmg.2005.034140. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous