

Core-shell nanoscale coordination polymers combine chemotherapy and photodynamic therapy to potentiate checkpoint blockade cancer immunotherapy
- PMID: 27530650
- PMCID: PMC4992065
- DOI: 10.1038/ncomms12499
Core-shell nanoscale coordination polymers combine chemotherapy and photodynamic therapy to potentiate checkpoint blockade cancer immunotherapy
Abstract
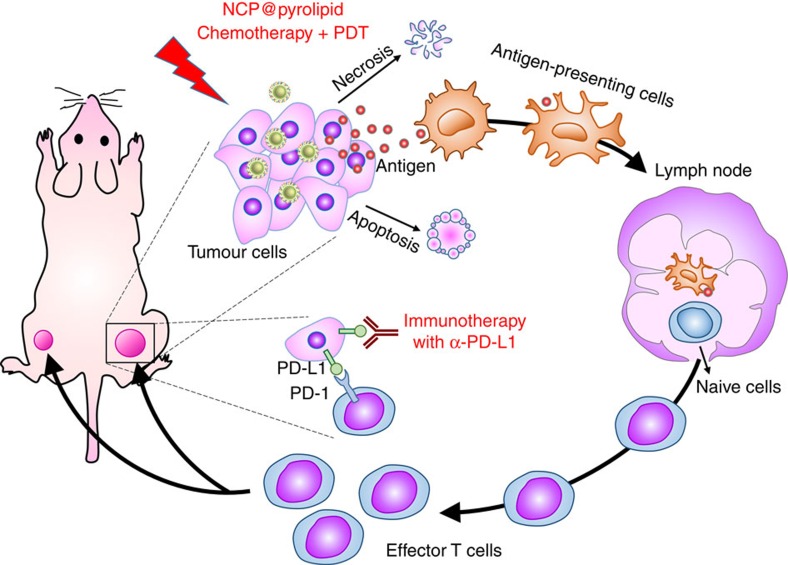
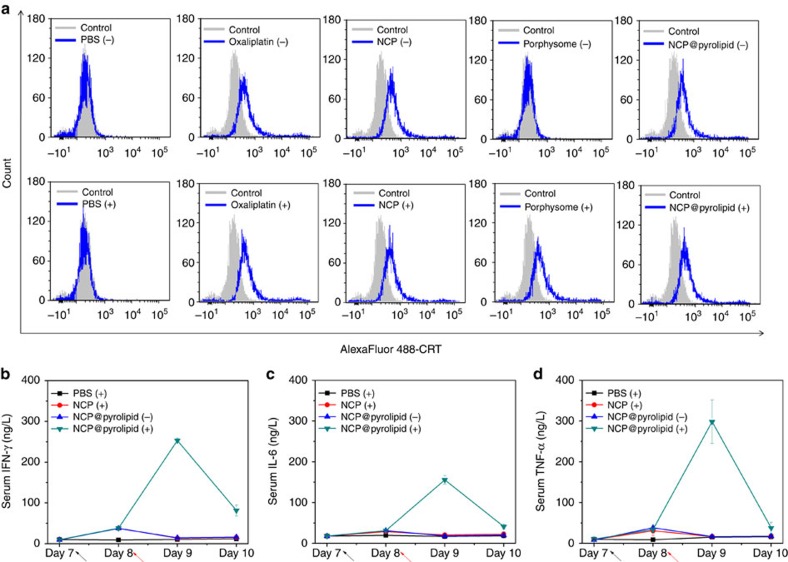
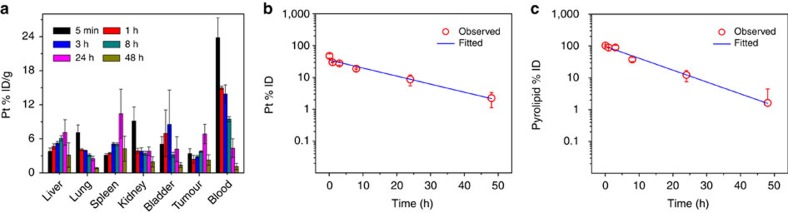
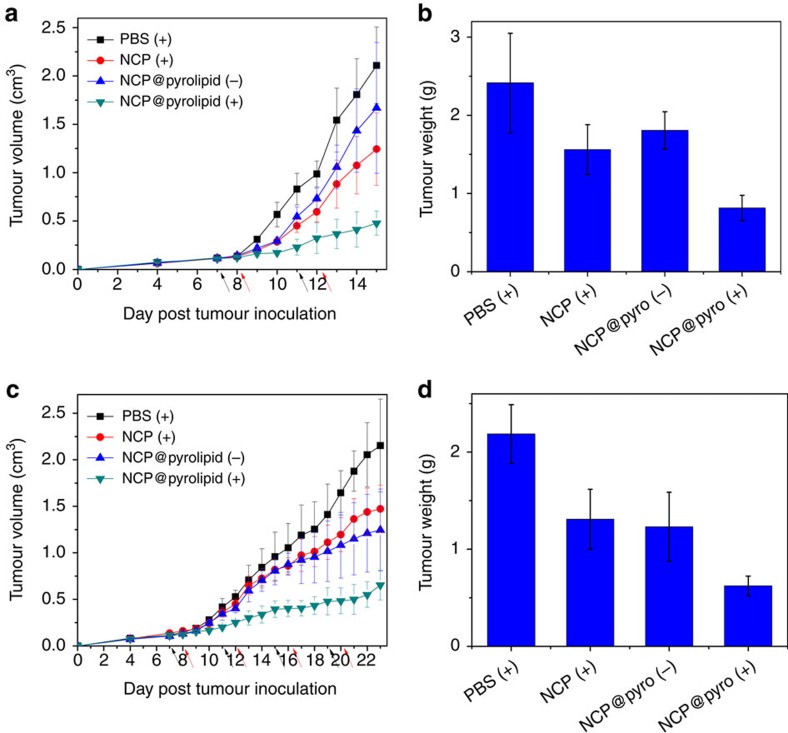
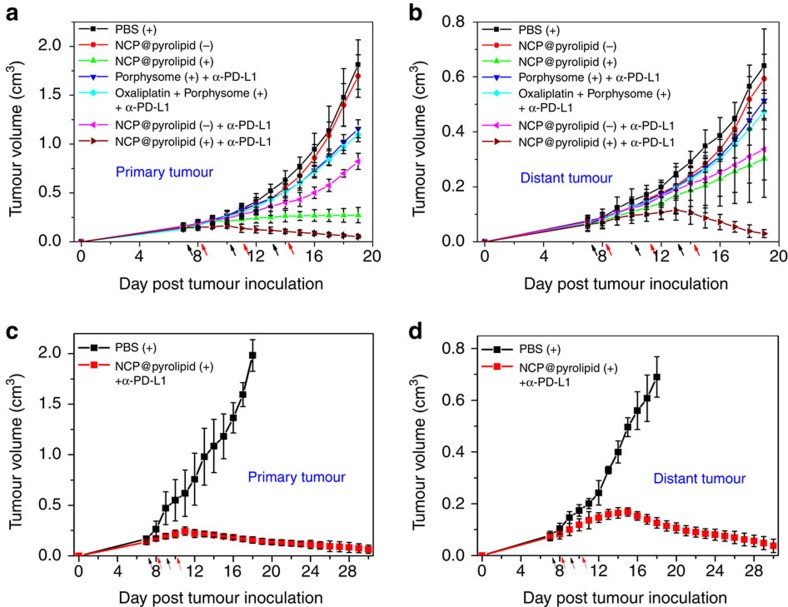
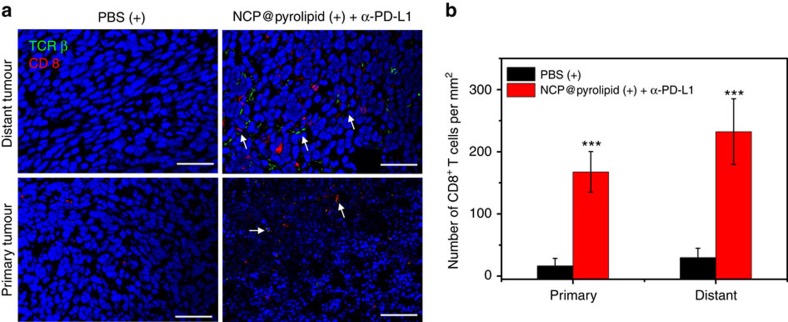
Advanced colorectal cancer is one of the deadliest cancers, with a 5-year survival rate of only 12% for patients with the metastatic disease. Checkpoint inhibitors, such as the antibodies inhibiting the PD-1/PD-L1 axis, are among the most promising immunotherapies for patients with advanced colon cancer, but their durable response rate remains low. We herein report the use of immunogenic nanoparticles to augment the antitumour efficacy of PD-L1 antibody-mediated cancer immunotherapy. Nanoscale coordination polymer (NCP) core-shell nanoparticles carry oxaliplatin in the core and the photosensitizer pyropheophorbide-lipid conjugate (pyrolipid) in the shell (NCP@pyrolipid) for effective chemotherapy and photodynamic therapy (PDT). Synergy between oxaliplatin and pyrolipid-induced PDT kills tumour cells and provokes an immune response, resulting in calreticulin exposure on the cell surface, antitumour vaccination and an abscopal effect. When combined with anti-PD-L1 therapy, NCP@pyrolipid mediates regression of both light-irradiated primary tumours and non-irradiated distant tumours by inducing a strong tumour-specific immune response.
Conflict of interest statement
W. L. is the founder of Coordination Pharmaceuticals, Inc., which licenses the NCP technology from the University of Chicago. R.R.W. is a consultant to Coordination Pharmaceuticals, Inc. All other authors declare no competing financial interest.
Figures


Comment in
-
Radiobiology and the Renewed Potential for Nanoparticles.Int J Radiat Oncol Biol Phys. 2017 Jul 1;98(3):489-491. doi: 10.1016/j.ijrobp.2017.03.015. Int J Radiat Oncol Biol Phys. 2017. PMID: 28581384 No abstract available.
Similar articles
-
Photodynamic Therapy Mediated by Nontoxic Core-Shell Nanoparticles Synergizes with Immune Checkpoint Blockade To Elicit Antitumor Immunity and Antimetastatic Effect on Breast Cancer.J Am Chem Soc. 2016 Dec 28;138(51):16686-16695. doi: 10.1021/jacs.6b09538. Epub 2016 Dec 15. J Am Chem Soc. 2016. PMID: 27976881 Free PMC article.
-
Self-assembled core-shell nanoparticles for combined chemotherapy and photodynamic therapy of resistant head and neck cancers.ACS Nano. 2015 Jan 27;9(1):991-1003. doi: 10.1021/nn506963h. Epub 2015 Jan 9. ACS Nano. 2015. PMID: 25559017
-
Immunostimulatory nanomedicines synergize with checkpoint blockade immunotherapy to eradicate colorectal tumors.Nat Commun. 2019 Apr 23;10(1):1899. doi: 10.1038/s41467-019-09221-x. Nat Commun. 2019. PMID: 31015397 Free PMC article.
-
Nanoparticle-based photothermal and photodynamic immunotherapy for tumor treatment.Int J Cancer. 2018 Dec 15;143(12):3050-3060. doi: 10.1002/ijc.31717. Epub 2018 Sep 27. Int J Cancer. 2018. PMID: 29981170 Review.
-
Immune-related adverse events with immune checkpoint blockade: a comprehensive review.Eur J Cancer. 2016 Feb;54:139-148. doi: 10.1016/j.ejca.2015.11.016. Epub 2016 Jan 5. Eur J Cancer. 2016. PMID: 26765102 Review.
Cited by
-
Quantum teleportation via a hybrid channel and investigation of its success probability.Sci Rep. 2024 Oct 29;14(1):26033. doi: 10.1038/s41598-024-76220-4. Sci Rep. 2024. PMID: 39472455 Free PMC article.
-
Engineering patient-specific cancer immunotherapies.Nat Biomed Eng. 2019 Oct;3(10):768-782. doi: 10.1038/s41551-019-0436-x. Epub 2019 Aug 12. Nat Biomed Eng. 2019. PMID: 31406259 Free PMC article. Review.
-
Immunophenotyping: Analytical approaches and role in preclinical development of nanomedicines.Adv Drug Deliv Rev. 2022 Jun;185:114281. doi: 10.1016/j.addr.2022.114281. Epub 2022 Apr 9. Adv Drug Deliv Rev. 2022. PMID: 35405297 Free PMC article. Review.
-
The Application of Nanoparticle-Based Drug Delivery Systems in Checkpoint Blockade Cancer Immunotherapy.J Immunol Res. 2018 Sep 30;2018:3673295. doi: 10.1155/2018/3673295. eCollection 2018. J Immunol Res. 2018. PMID: 30406152 Free PMC article. Review.
-
Immunogenic Cell Death Induced by Chemoradiotherapy of Novel pH-Sensitive Cargo-Loaded Polymersomes in Glioblastoma.Int J Nanomedicine. 2021 Oct 22;16:7123-7135. doi: 10.2147/IJN.S333197. eCollection 2021. Int J Nanomedicine. 2021. PMID: 34712045 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
