

Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples
- PMID: 27532257
- PMCID: PMC5116235
- DOI: 10.1038/gim.2016.90
Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples
Abstract
Purpose: The accurate interpretation of variation in Mendelian disease genes has lagged behind data generation as sequencing has become increasingly accessible. Ongoing large sequencing efforts present huge interpretive challenges, but they also provide an invaluable opportunity to characterize the spectrum and importance of rare variation.
Methods: We analyzed sequence data from 7,855 clinical cardiomyopathy cases and 60,706 Exome Aggregation Consortium (ExAC) reference samples to obtain a better understanding of genetic variation in a representative autosomal dominant disorder.
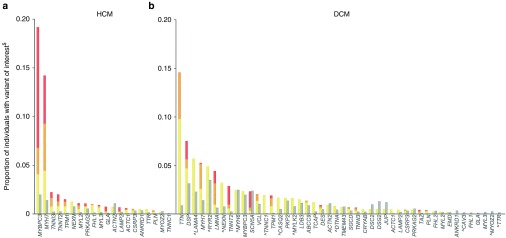
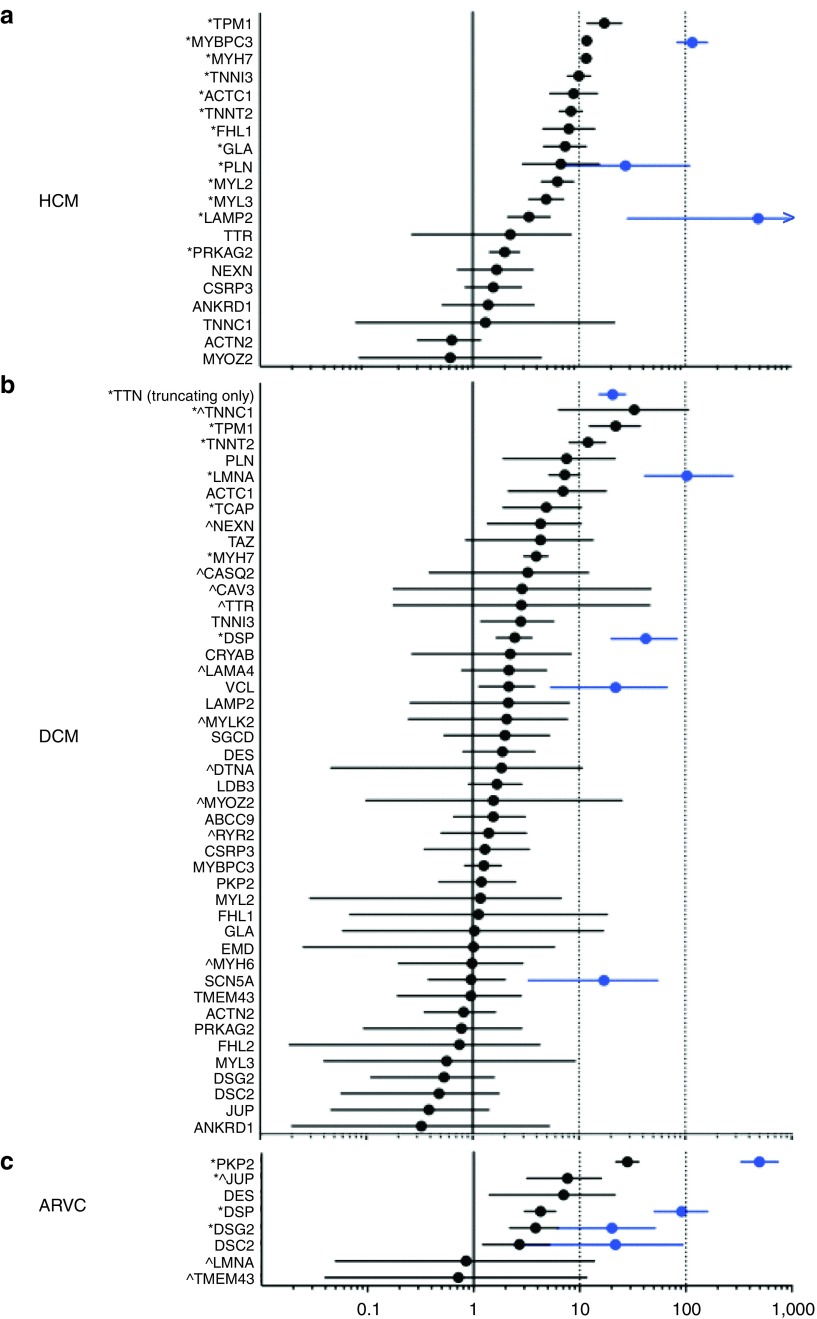
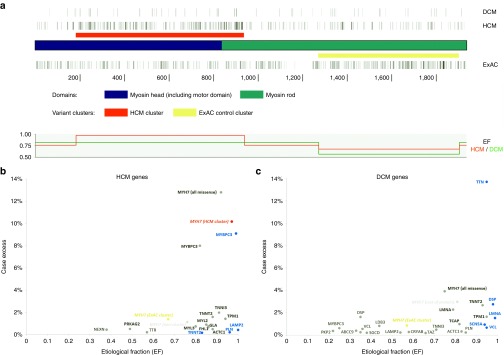
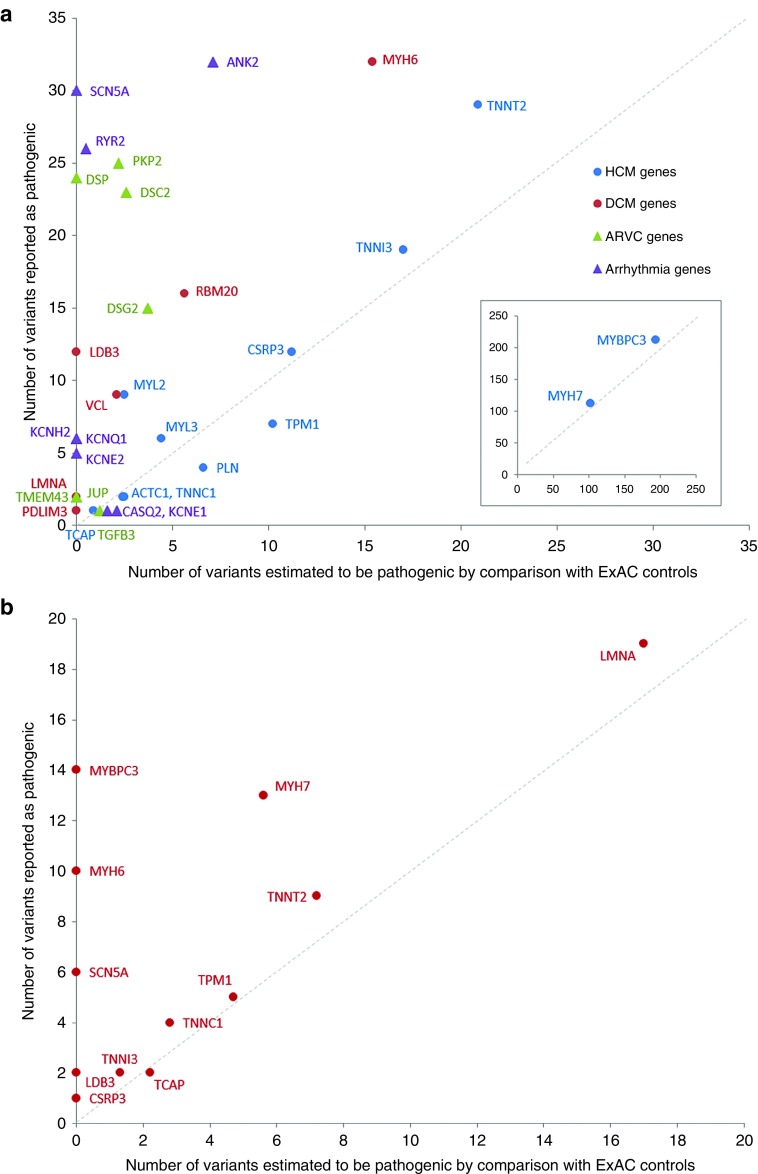
Results: We found that in some genes previously reported as important causes of a given cardiomyopathy, rare variation is not clinically informative because there is an unacceptably high likelihood of false-positive interpretation. By contrast, in other genes, we find that diagnostic laboratories may be overly conservative when assessing variant pathogenicity.
Conclusions: We outline improved analytical approaches that evaluate which genes and variant classes are interpretable and propose that these will increase the clinical utility of testing across a range of Mendelian diseases.Genet Med 19 2, 192-203.
Conflict of interest statement
None
Figures
References
-
- Xue Y, Ankala A, Wilcox WR, Hegde MR. Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing: single-gene, gene panel, or exome/genome sequencing. Genet Med 2015;17:444–451. - PubMed
-
- Wooderchak-Donahue W, VanSant-Webb C, Tvrdik T, et al. Clinical utility of a next generation sequencing panel assay for Marfan and Marfan-like syndromes featuring aortopathy. Am J Med Genet Part A 2015;167:1747–1757. - PubMed
-
- Pugh TJ, Kelly MA, Gowrisankar S, et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med 2014;16:601–608. - PubMed
-
- Exome Aggregation Consortium; Lek M, Karczewski K, Minikel E, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature, e-pub ahead of print 17 August, 2016. http://biorxiv.org/content/early/2015/10/30/030338. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
