

Complement-Mediated Regulation of Metabolism and Basic Cellular Processes
- PMID: 27533012
- PMCID: PMC5019180
- DOI: 10.1016/j.immuni.2016.08.003
Complement-Mediated Regulation of Metabolism and Basic Cellular Processes
Abstract
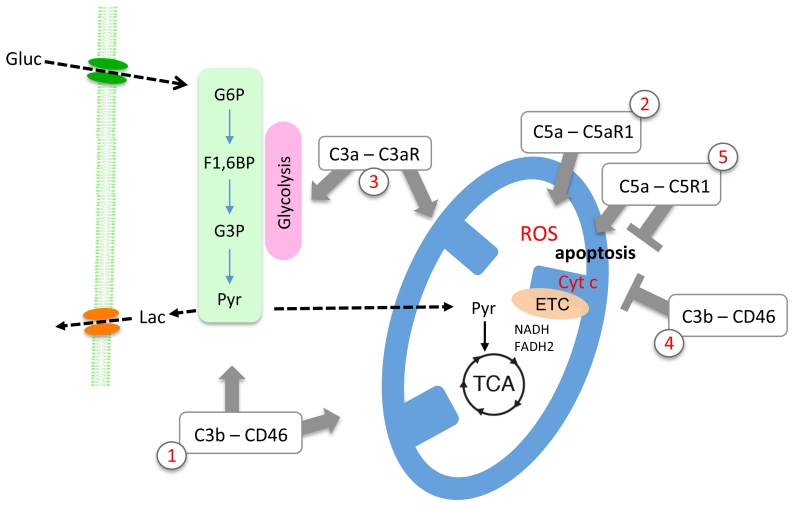
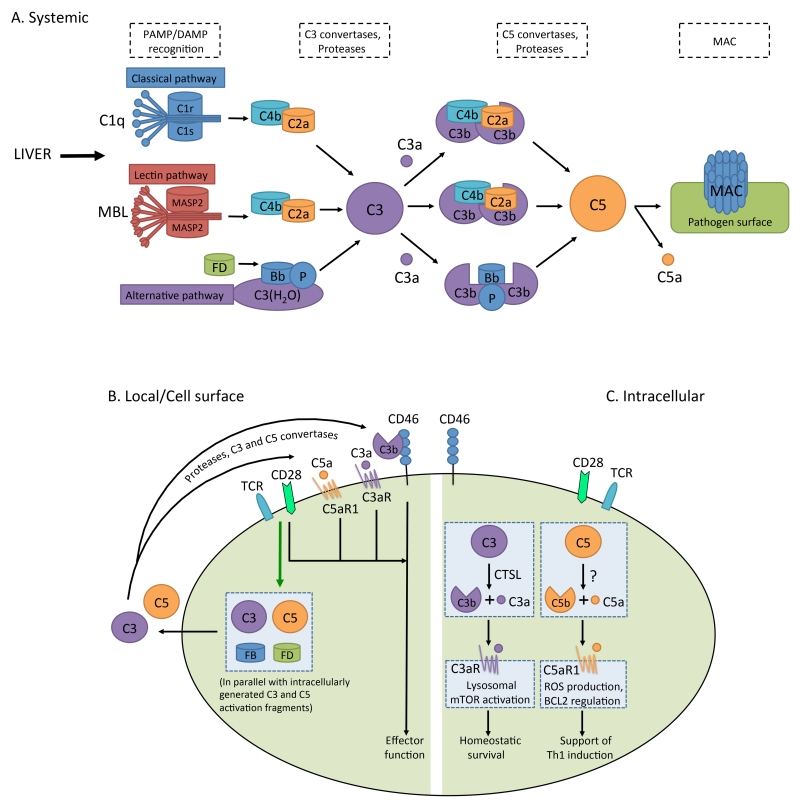
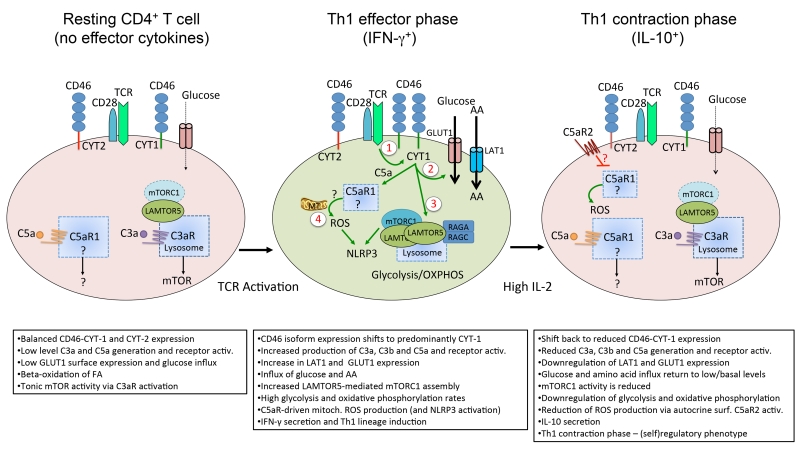
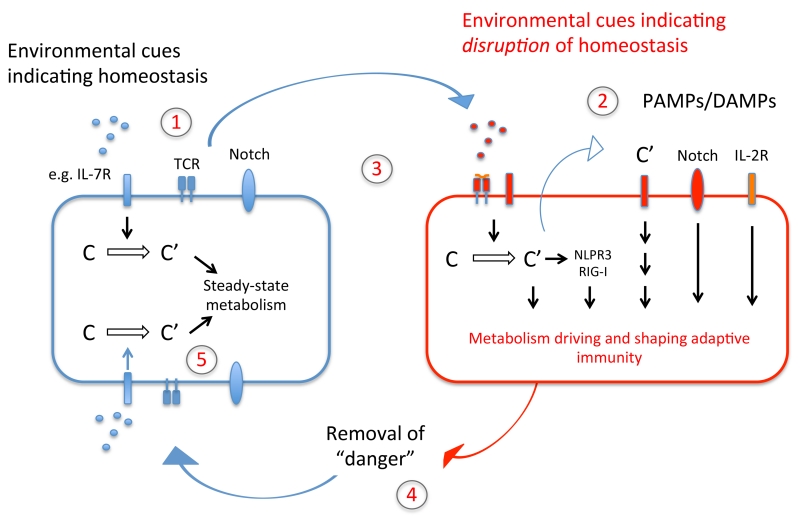
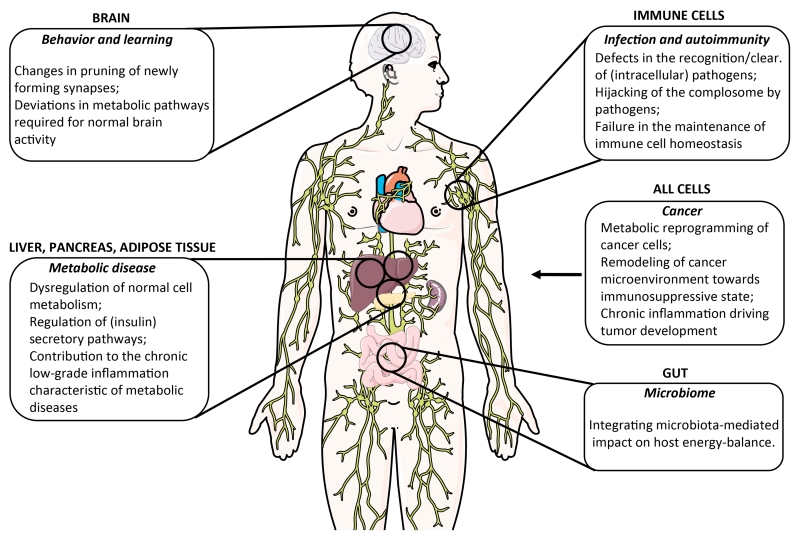
Complement is well appreciated as a critical arm of innate immunity. It is required for the removal of invading pathogens and works by directly destroying them through the activation of innate and adaptive immune cells. However, complement activation and function is not confined to the extracellular space but also occurs within cells. Recent work indicates that complement activation regulates key metabolic pathways and thus can impact fundamental cellular processes, such as survival, proliferation, and autophagy. Newly identified functions of complement include a key role in shaping metabolic reprogramming, which underlies T cell effector differentiation, and a role as a nexus for interactions with other effector systems, in particular the inflammasome and Notch transcription-factor networks. This review focuses on the contributions of complement to basic processes of the cell, in particular the integration of complement with cellular metabolism and the potential implications in infection and other disease settings.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures
References
-
- Alexander JJ, Jacob A, Bao L, Macdonald RL, Quigg RJ. Complement-dependent apoptosis and inflammatory gene changes in murine lupus cerebritis. J Immunol. 2005;175:8312–8319. - PubMed
-
- Amsen D, Helbig C, Backer RA. Notch in T Cell Differentiation: All Things Considered. Trends Immunol. 2015;36:802–814. - PubMed
-
- Asgari E, Le Friec G, Yamamoto H, Perucha E, Sacks SS, Kohl J, Cook HT, Kemper C. C3a modulates IL-1beta secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood. 2013;122:3473–3481. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
