

Structure-based discovery of opioid analgesics with reduced side effects
- PMID: 27533032
- PMCID: PMC5161585
- DOI: 10.1038/nature19112
Structure-based discovery of opioid analgesics with reduced side effects
Abstract
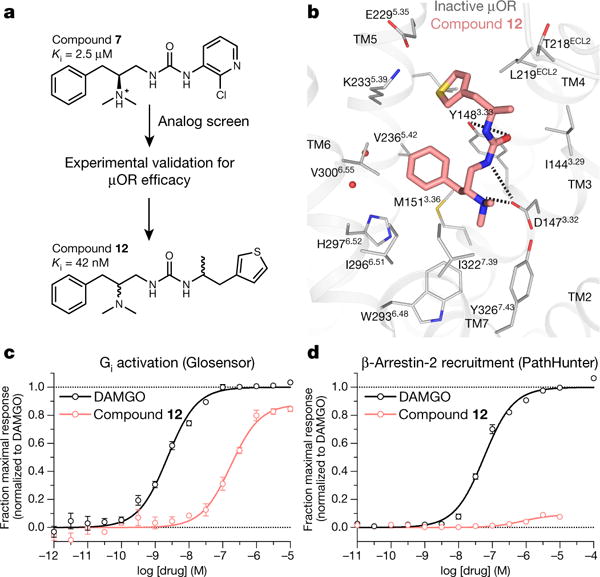
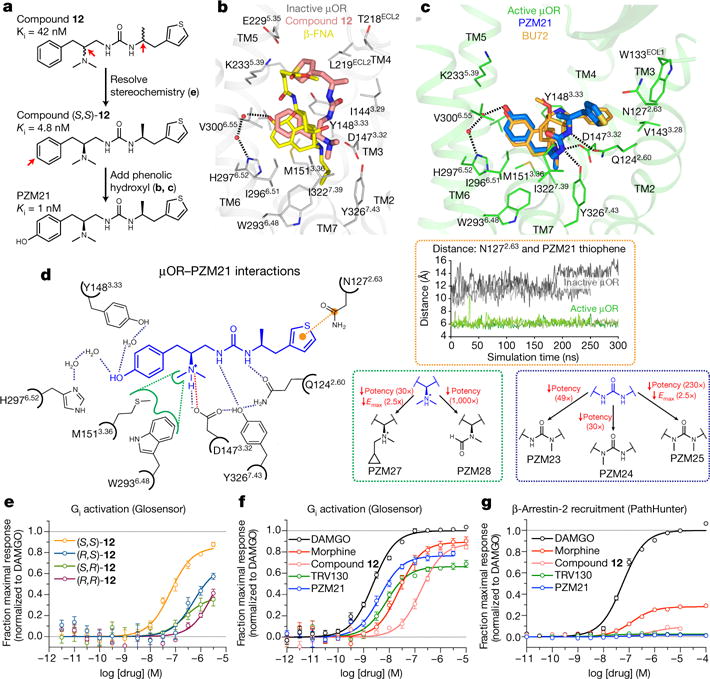
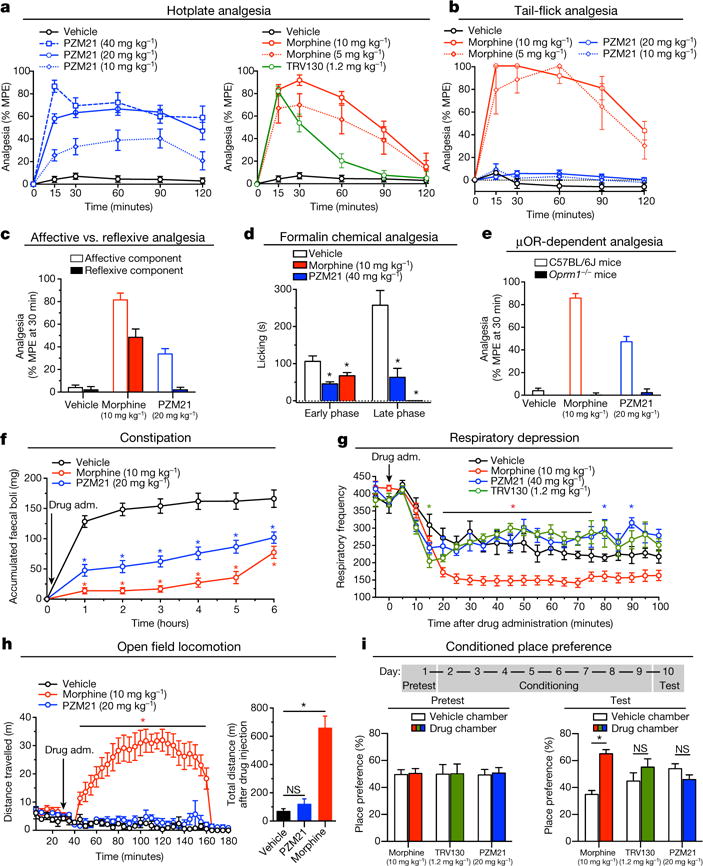
Morphine is an alkaloid from the opium poppy used to treat pain. The potentially lethal side effects of morphine and related opioids-which include fatal respiratory depression-are thought to be mediated by μ-opioid-receptor (μOR) signalling through the β-arrestin pathway or by actions at other receptors. Conversely, G-protein μOR signalling is thought to confer analgesia. Here we computationally dock over 3 million molecules against the μOR structure and identify new scaffolds unrelated to known opioids. Structure-based optimization yields PZM21-a potent Gi activator with exceptional selectivity for μOR and minimal β-arrestin-2 recruitment. Unlike morphine, PZM21 is more efficacious for the affective component of analgesia versus the reflexive component and is devoid of both respiratory depression and morphine-like reinforcing activity in mice at equi-analgesic doses. PZM21 thus serves as both a probe to disentangle μOR signalling and a therapeutic lead that is devoid of many of the side effects of current opioids.
Conflict of interest statement
The authors declare competing financial interests: details are available in the online version of the paper. Readers are welcome to comment on the online version of the paper.
Figures
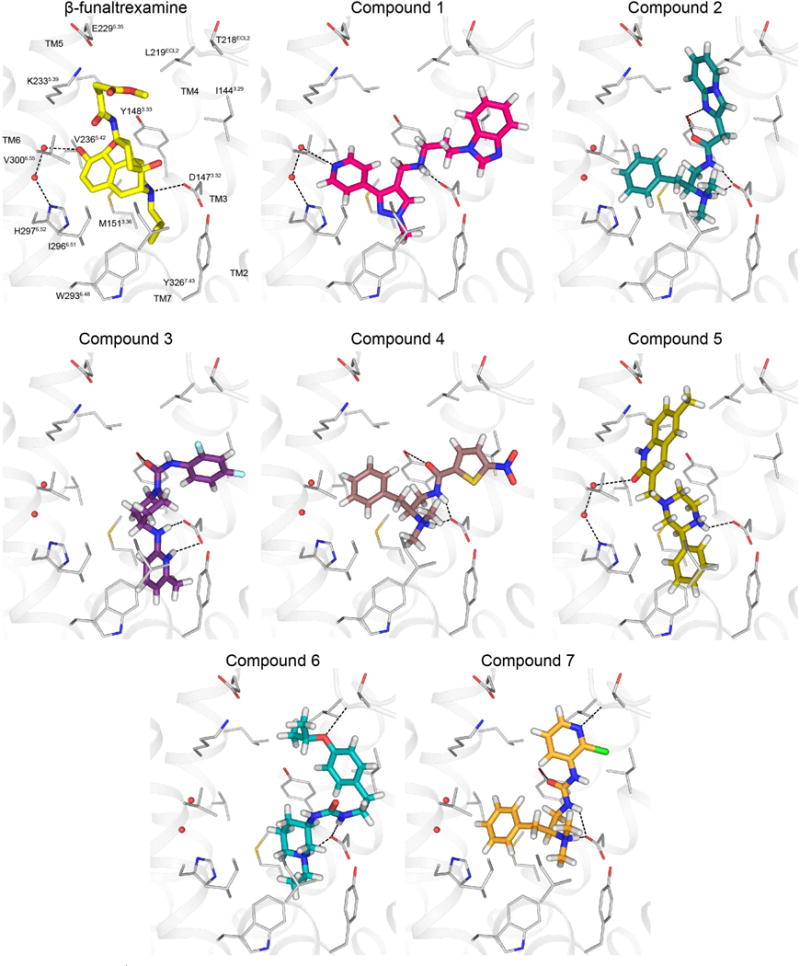
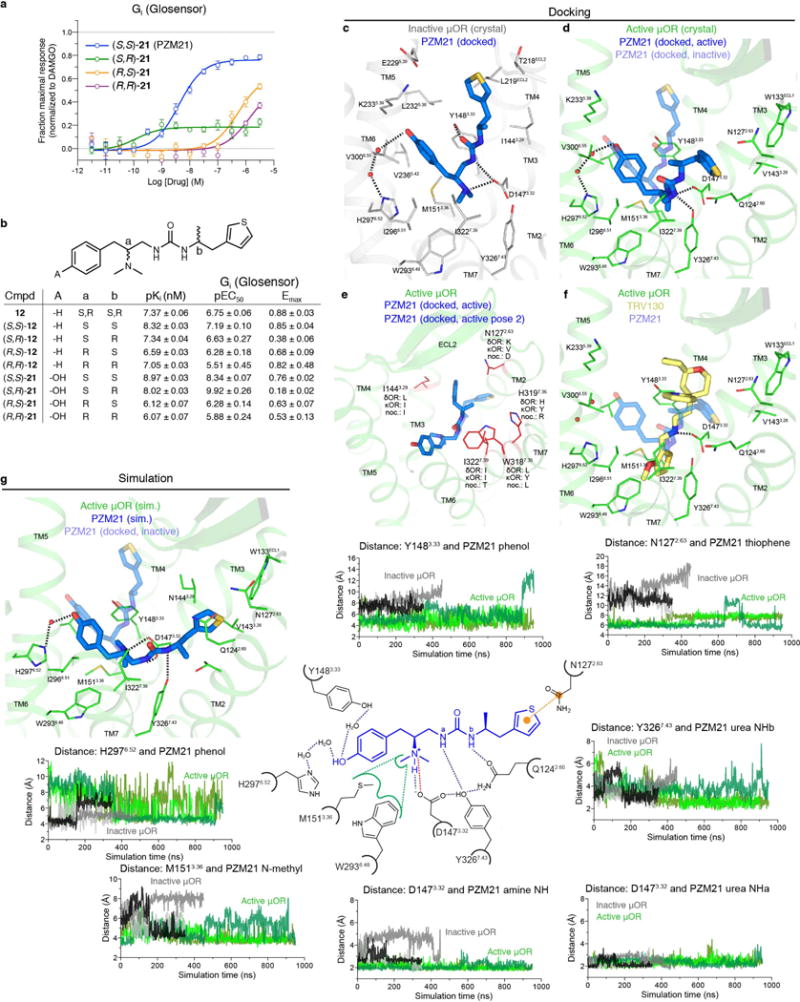
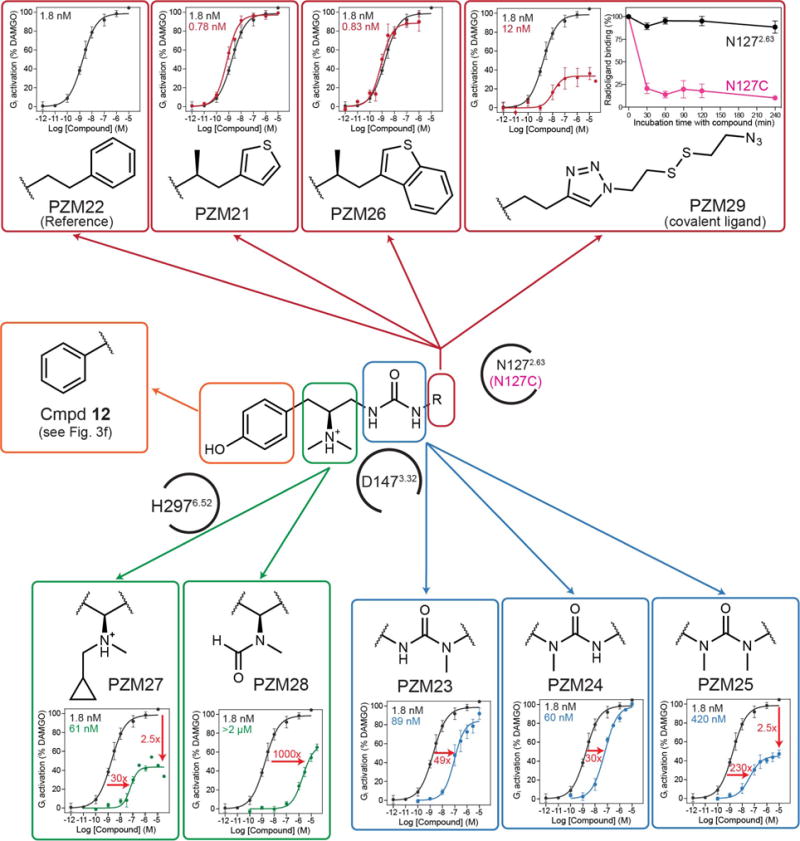
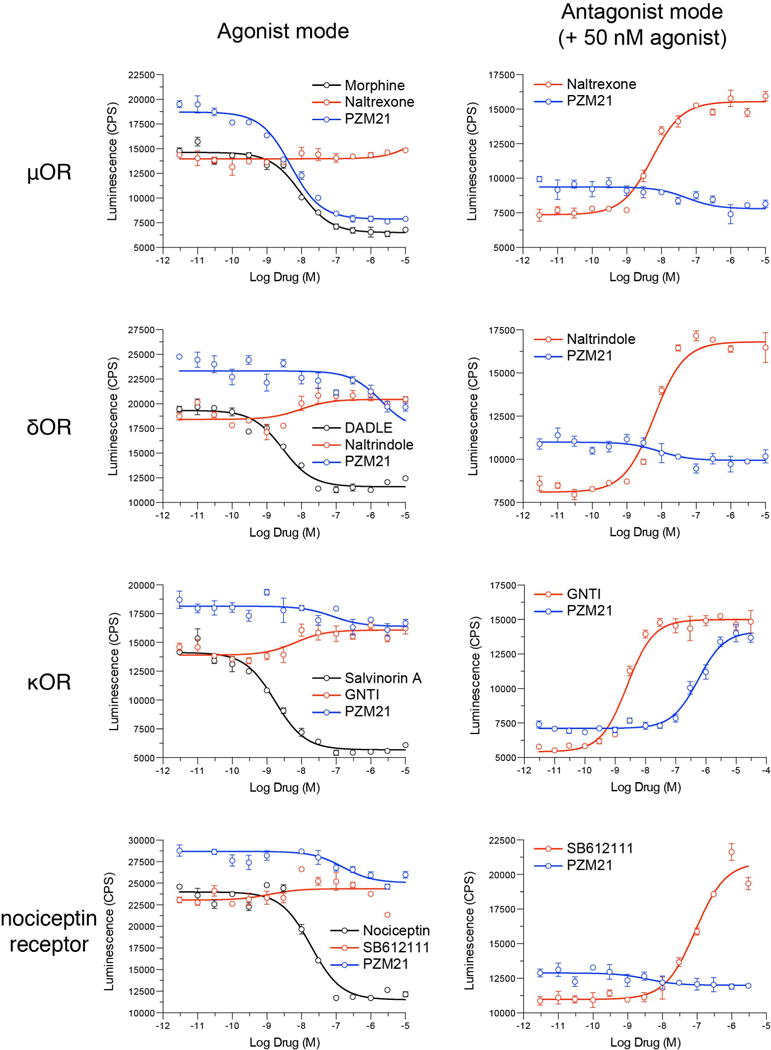
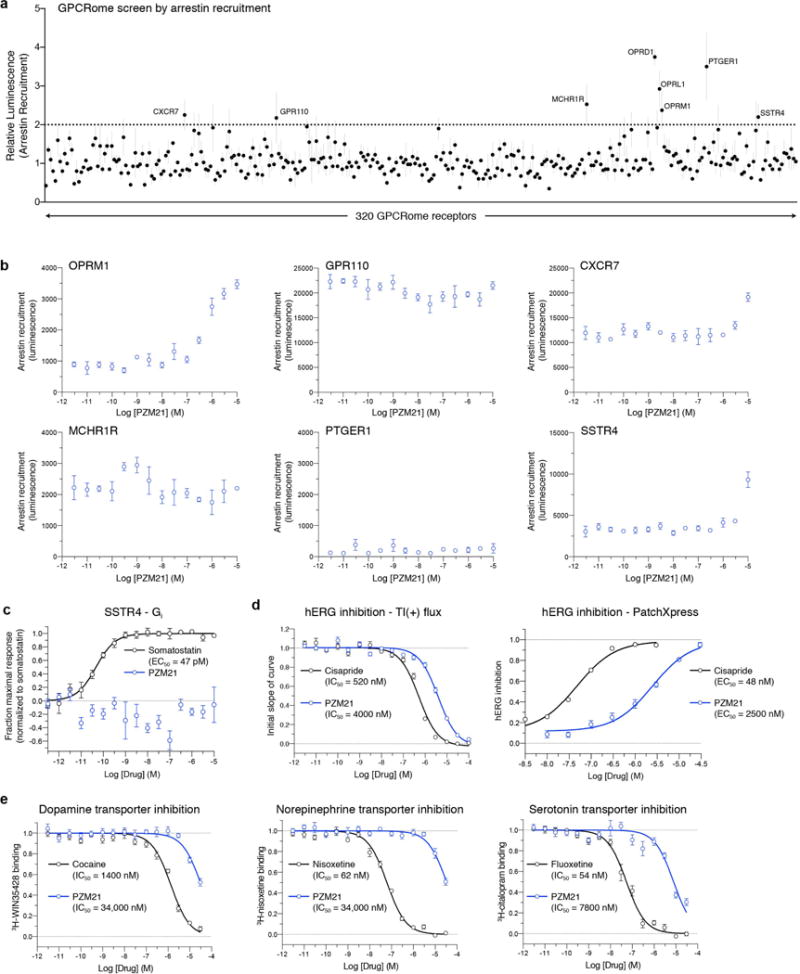
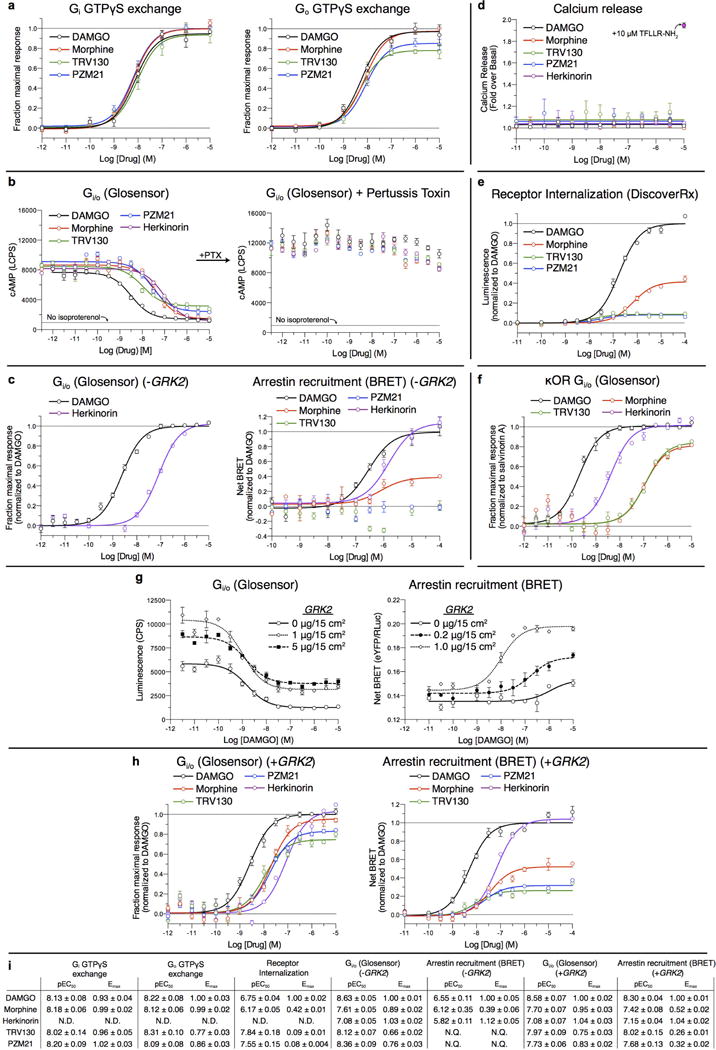
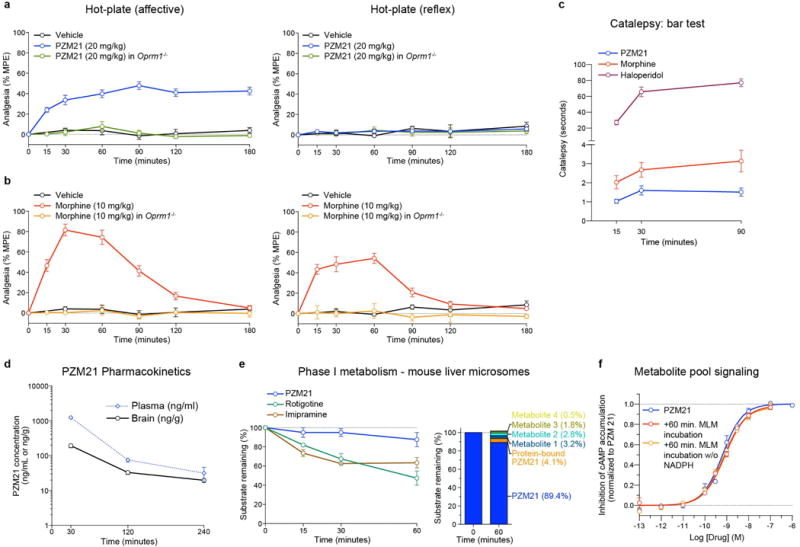
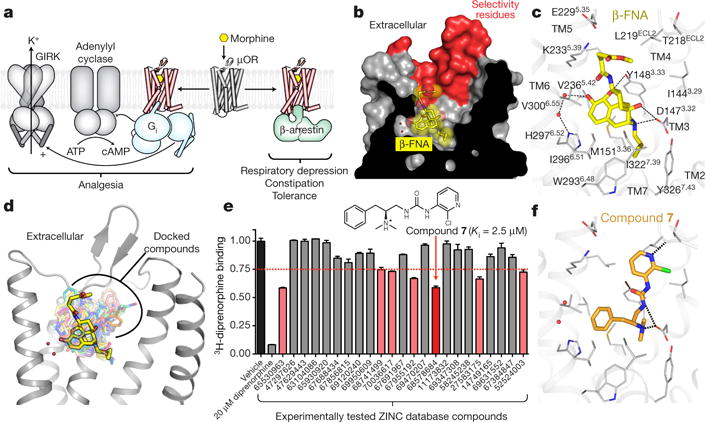
Comment in
-
Drug discovery: Designing the ideal opioid.Nature. 2016 Sep 8;537(7619):170-171. doi: 10.1038/nature19424. Epub 2016 Aug 17. Nature. 2016. PMID: 27533037 No abstract available.
References
-
- Lord JAH, Waterfield AA, Hughes J, Kosterlitz HW. Endogenous opioid peptides: multiple agonists and receptors. Nature. 1977;267:495–499. - PubMed
-
- Martin WR, Eades CG, Thompson JA, Huppler RE, Gilbert PE. The effects of morphine- and nalorphine-like drugs in the nondependent and morphine-dependent chronic spinal dog. J Pharmacol Exp Ther. 1976;197:517–532. - PubMed
-
- Hughes J, et al. Identification of two related pentapeptides from the brain with potent opiate agonist activity. Nature. 1975;258:577–580. - PubMed
-
- Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. μ-opioid receptor desensitization by β-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408:720–723. - PubMed
-
- Bohn LM, et al. Enhanced morphine analgesia in mice lacking β-arrestin 2. Science. 1999;286:2495–2498. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
