

Dysregulation of Neuronal Ca2+ Channel Linked to Heightened Sympathetic Phenotype in Prohypertensive States
- PMID: 27535905
- PMCID: PMC4987433
- DOI: 10.1523/JNEUROSCI.1059-16.2016
Dysregulation of Neuronal Ca2+ Channel Linked to Heightened Sympathetic Phenotype in Prohypertensive States
Abstract
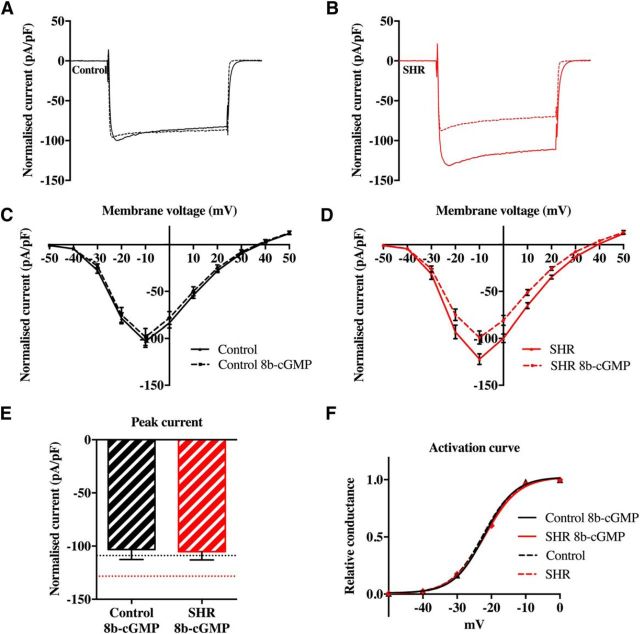
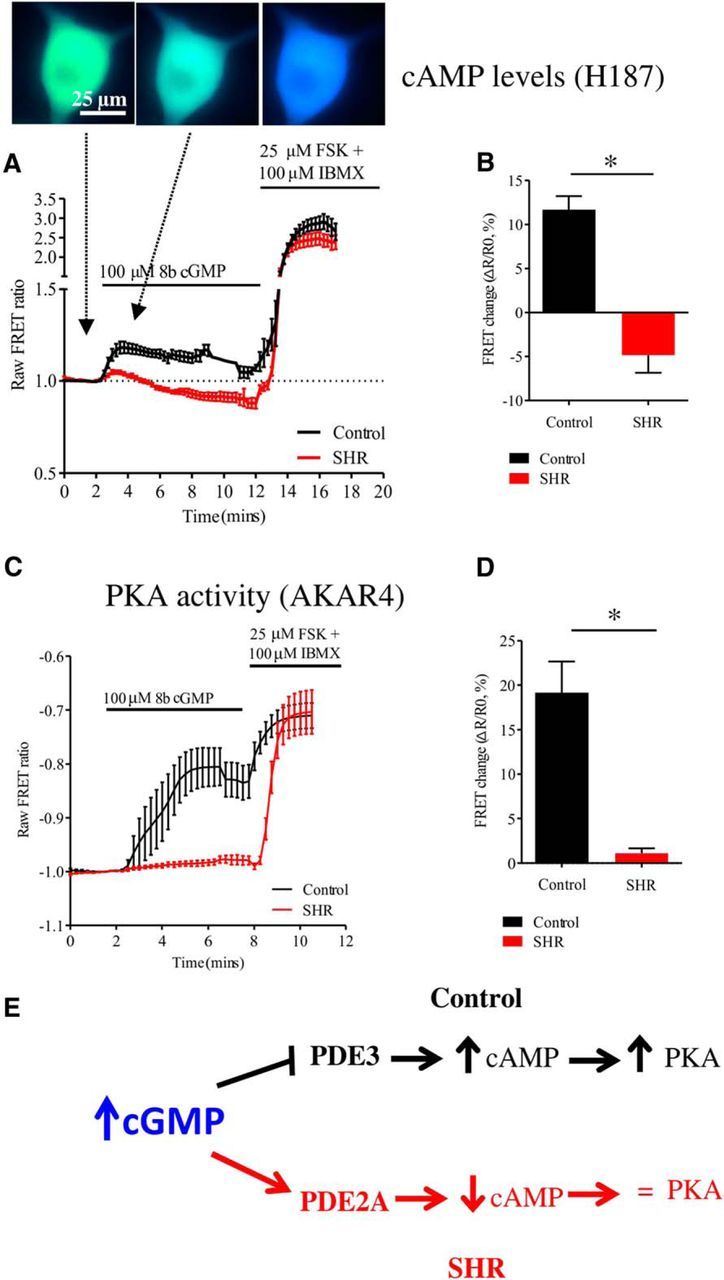
Hypertension is associated with impaired nitric oxide (NO)-cyclic nucleotide (CN)-coupled intracellular calcium (Ca(2+)) homeostasis that enhances cardiac sympathetic neurotransmission. Because neuronal membrane Ca(2+) currents are reduced by NO-activated S-nitrosylation, we tested whether CNs affect membrane channel conductance directly in neurons isolated from the stellate ganglia of spontaneously hypertensive rats (SHRs) and their normotensive controls. Using voltage-clamp and cAMP-protein kinase A (PKA) FRET sensors, we hypothesized that impaired CN regulation provides a direct link to abnormal signaling of neuronal calcium channels in the SHR and that targeting cGMP can restore the channel phenotype. We found significantly larger whole-cell Ca(2+) currents from diseased neurons that were largely mediated by the N-type Ca(2+) channel (Cav2.2). Elevating cGMP restored the SHR Ca(2+) current to levels seen in normal neurons that were not affected by cGMP. cGMP also decreased cAMP levels and PKA activity in diseased neurons. In contrast, cAMP-PKA activity was increased in normal neurons, suggesting differential switching in phosphodiesterase (PDE) activity. PDE2A inhibition enhanced the Ca(2+) current in normal neurons to a conductance similar to that seen in SHR neurons, whereas the inhibitor slightly decreased the current in diseased neurons. Pharmacological evidence supported a switching from cGMP acting via PDE3 in control neurons to PDE2A in SHR neurons in the modulation of the Ca(2+) current. Our data suggest that a disturbance in the regulation of PDE-coupled CNs linked to N-type Ca(2+) channels is an early hallmark of the prohypertensive phenotype associated with intracellular Ca(2+) impairment underpinning sympathetic dysautonomia.
Significance statement: Here, we identify dysregulation of cyclic-nucleotide (CN)-linked neuronal Ca(2+) channel activity that could provide the trigger for the enhanced sympathetic neurotransmission observed in the prohypertensive state. Furthermore, we provide evidence that increasing cGMP rescues the channel phenotype and restores ion channel activity to levels seen in normal neurons. We also observed CN cross-talk in sympathetic neurons that may be related to a differential switching in phosphodiesterase activity. The presence of these early molecular changes in asymptomatic, prohypertensive animals could facilitate the identification of novel therapeutic targets with which to modulate intracellular Ca(2+) Turning down the gain of sympathetic hyperresponsiveness in cardiovascular disease associated with sympathetic dysautonomia would have significant therapeutic utility.
Keywords: autonomic nervous system; cGMP; dysautonomia; hypertension; N-type Ca2+ channel; sympathetic.
Copyright © 2016 Larsen et al.
Figures






Comment in
-
Impaired cAMP-cGMP cross-talk during cardiac sympathetic dysautonomia.Channels (Austin). 2017 May 4;11(3):178-180. doi: 10.1080/19336950.2016.1259040. Epub 2016 Nov 11. Channels (Austin). 2017. PMID: 27835060 Free PMC article. No abstract available.
References
-
- Bannister RA, Meza U, Adams BA. Phosphorylation-dependent regulation of voltage-gated Ca2+ channels. In: Zamponi G., editor. Voltage-gated calcium channels. Boston: Springer; 2005. pp. 168–182.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous