

The Prevalence and Management of Systemic Amyloidosis in Western Countries
- PMID: 27536687
- PMCID: PMC4946260
- DOI: 10.1159/000444206
The Prevalence and Management of Systemic Amyloidosis in Western Countries
Abstract
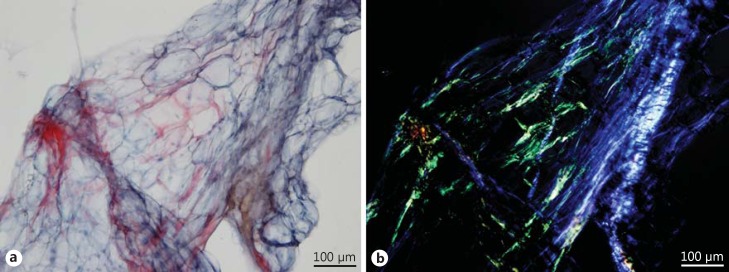
Background: Amyloidosis has been a mystery for centuries, but research of the last decennia has clarified many of the secrets of this group of diseases. A protein-based classification of amyloidosis helps to understand problems that were part of the obsolete clinical classification in primary, secondary, and familial amyloidosis. All types of amyloid are secondary to some underlying precursor-producing process: each type is caused by a misfolded soluble precursor protein that becomes deposited as insoluble amyloid fibrils.
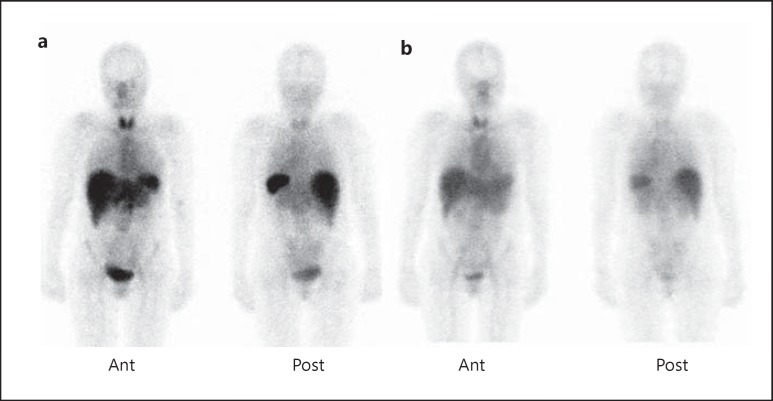
Summary: The incidence of amyloidosis is not well documented, but probably falls between 5 and 13 per million per year. Prevalence data are scarce, but one UK study indicates about 20 per million inhabitants. Amyloidosis can be localized (amyloid deposited in the organ or tissue of precursor production) or systemic (amyloid at one or more sites distant from the site of precursor production). The major systemic types of amyloidosis are AL (associated with a light chain-producing plasma cell dyscrasia), AA (associated with longstanding inflammation), wild-type ATTR (associated with normal transthyretin and old age), and hereditary ATTR (associated with a transthyretin mutation) amyloidosis. Imaging techniques, such as cardiac ultrasound, magnetic resonance imaging, bone scintigraphy, and serum amyloid P component scintigraphy, are useful both for diagnosing amyloidosis and for assessing disease severity. Serologic markers are useful for detecting organ disease and disease monitoring during follow-up. Current treatment modalities are directed against the ongoing supply of precursor proteins and thereby aim to stop further accumulation of amyloid. Novel treatment modalities, such as interference with amyloid formation and even removal of amyloid, are being studied. A well-thought and planned monitoring during follow-up helps to assess the effect of treatment and to early detect possible progression of amyloidosis.
Key messages: Clinical management comprises histologic proof of amyloid, evidence of systemic deposition, reliable typing, precursor assessment, severity of organ disease, risk assessment and prognosis, choice of treatment, and planned monitoring during follow-up.
Facts from east and west: (1) AL amyloidosis is the most prevalent type of amyloidosis accounting for 65% of the amyloidosis-diagnosed patients in the UK and for 93% of the amyloidosis-diagnosed patients in China. The predisposition of men over women to develop AL amyloidosis might be higher in China than in Western countries (2:1 vs. 1.3:1). Both in the East and West, incidence increases with age. At the time of diagnosis, edema is twice as frequent and the proportion of renal involvement is higher in Chinese compared to Western patients. (2) Melphalan followed by autologous stem cell transplantation (ASCT) is the current standard therapy but is restricted to eligible patients. The efficacy and safety of bortezomib combined with dexamethasone were proven in Western patients and recently confirmed in a Chinese cohort. Recent studies in China and the US indicate that bortezomib induction prior to ASCT increases the response rate. Thalidomide and lenalidomide have shown benefit, but toxicity and lack of clinical evidence exclude these agents from first-line therapy. The green tea extract epigallocatechin-3-gallate is under investigation as an inhibitor of AL amyloid formation and a compound that might dissolve amyloid.
Keywords: Diagnosis; Monitoring; Precursor proteins; Treatment; Typing.
Figures

References
-
- Kyle RA. Amyloidosis: a convoluted story. Br J Haematol. 2001;114:529–538. - PubMed
-
- Sipe JD, Benson MD, Buxbaum JN, Ikeda S, Merlini G, Saraiva MJ, Westermark P. Nomenclature 2014: amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid. 2014;21:221–224. - PubMed
-
- Kyle RA, Linos A, Beard CM, Linke RP, Gertz MA, O'Fallon WM, Kurland LT. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood. 1992;79:1817–1822. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
