

A Genomic Map of the Effects of Linked Selection in Drosophila
- PMID: 27536991
- PMCID: PMC4990265
- DOI: 10.1371/journal.pgen.1006130
A Genomic Map of the Effects of Linked Selection in Drosophila
Abstract
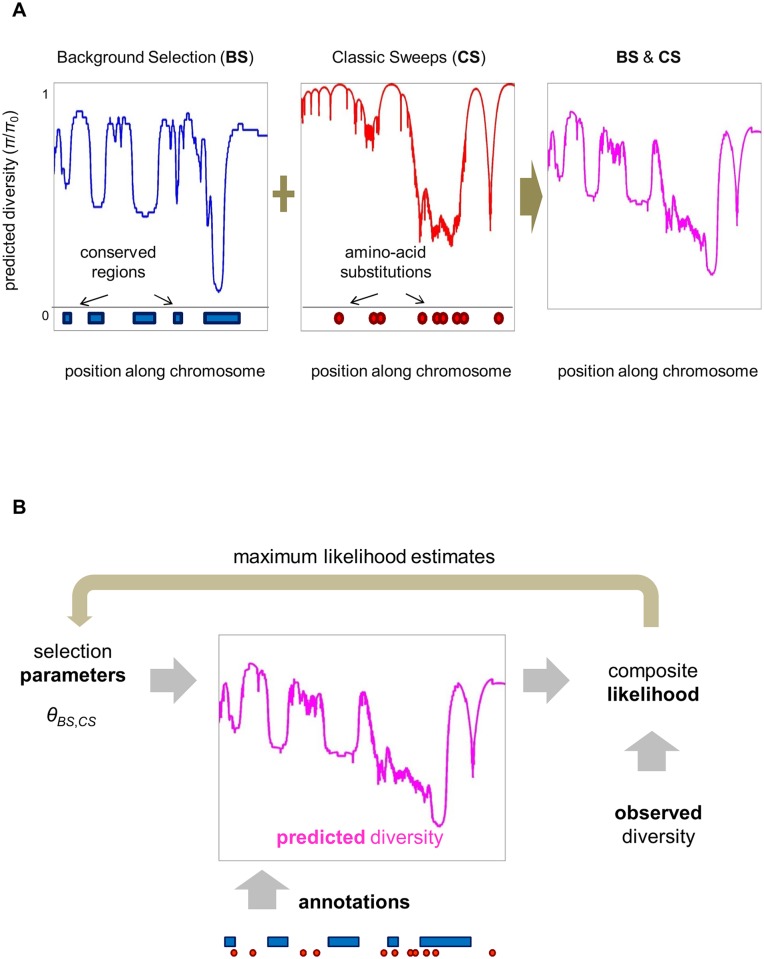
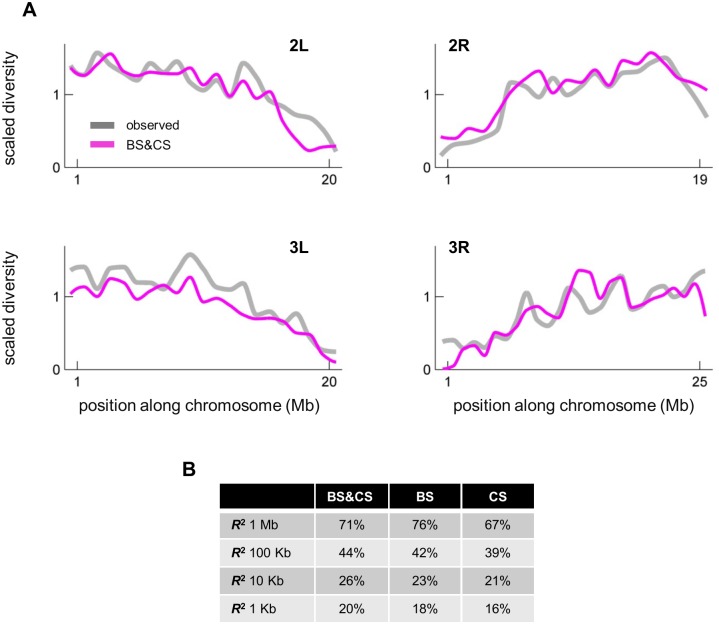
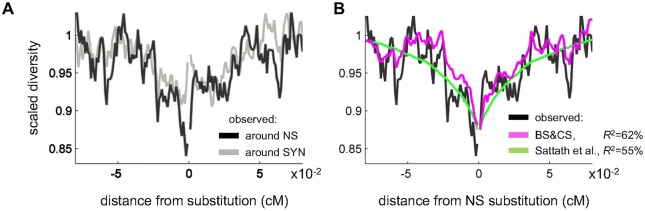
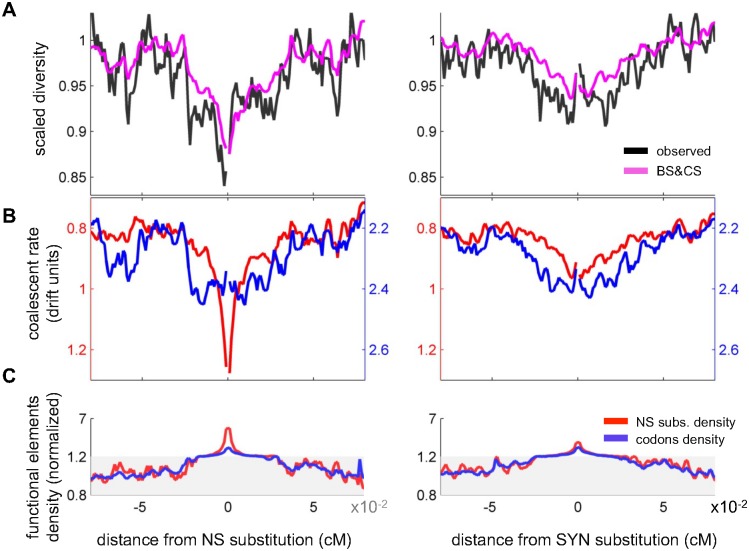
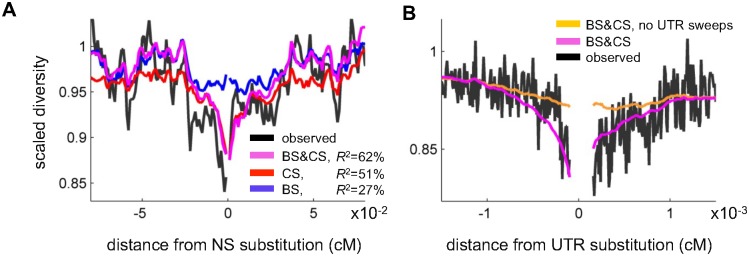
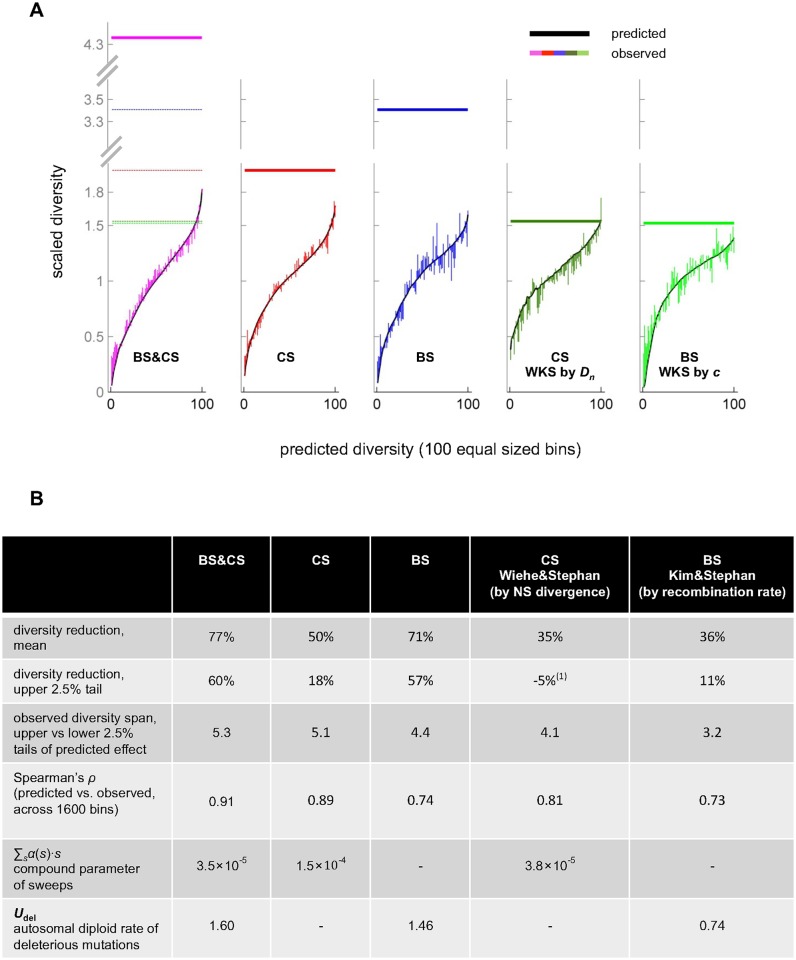
Natural selection at one site shapes patterns of genetic variation at linked sites. Quantifying the effects of "linked selection" on levels of genetic diversity is key to making reliable inference about demography, building a null model in scans for targets of adaptation, and learning about the dynamics of natural selection. Here, we introduce the first method that jointly infers parameters of distinct modes of linked selection, notably background selection and selective sweeps, from genome-wide diversity data, functional annotations and genetic maps. The central idea is to calculate the probability that a neutral site is polymorphic given local annotations, substitution patterns, and recombination rates. Information is then combined across sites and samples using composite likelihood in order to estimate genome-wide parameters of distinct modes of selection. In addition to parameter estimation, this approach yields a map of the expected neutral diversity levels along the genome. To illustrate the utility of our approach, we apply it to genome-wide resequencing data from 125 lines in Drosophila melanogaster and reliably predict diversity levels at the 1Mb scale. Our results corroborate estimates of a high fraction of beneficial substitutions in proteins and untranslated regions (UTR). They allow us to distinguish between the contribution of sweeps and other modes of selection around amino acid substitutions and to uncover evidence for pervasive sweeps in untranslated regions (UTRs). Our inference further suggests a substantial effect of other modes of linked selection and of adaptation in particular. More generally, we demonstrate that linked selection has had a larger effect in reducing diversity levels and increasing their variance in D. melanogaster than previously appreciated.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Comment in
-
On the Trail of Linked Selection.PLoS Genet. 2016 Aug 18;12(8):e1006240. doi: 10.1371/journal.pgen.1006240. eCollection 2016 Aug. PLoS Genet. 2016. PMID: 27537331 Free PMC article. No abstract available.
References
-
- Hill WG, Robertson A (1966) The effect of linkage on limits to artificial selection. Genet Res 8: 269–294. - PubMed
-
- Maynard Smith JM, Haigh J (1974) The hitch-hiking effect of a favourable gene. Genet Res 23: 23–35. - PubMed
-
- Begun DJ, Aquadro CF (1992) Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster. Nature 356: 519–520. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
