

Tumor Protein (TP)-p53 Members as Regulators of Autophagy in Tumor Cells upon Marine Drug Exposure
- PMID: 27537898
- PMCID: PMC4999915
- DOI: 10.3390/md14080154
Tumor Protein (TP)-p53 Members as Regulators of Autophagy in Tumor Cells upon Marine Drug Exposure
Abstract
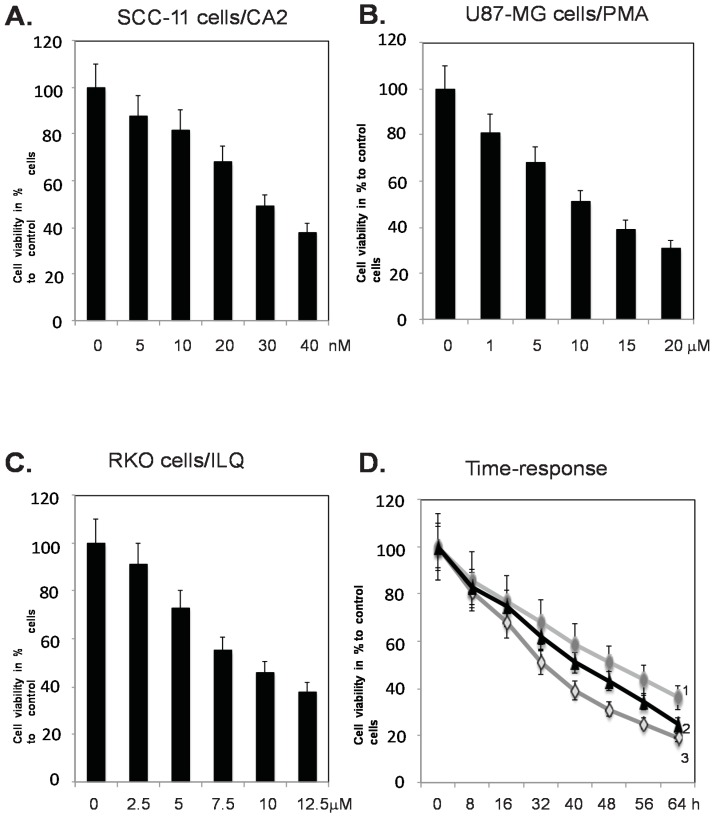
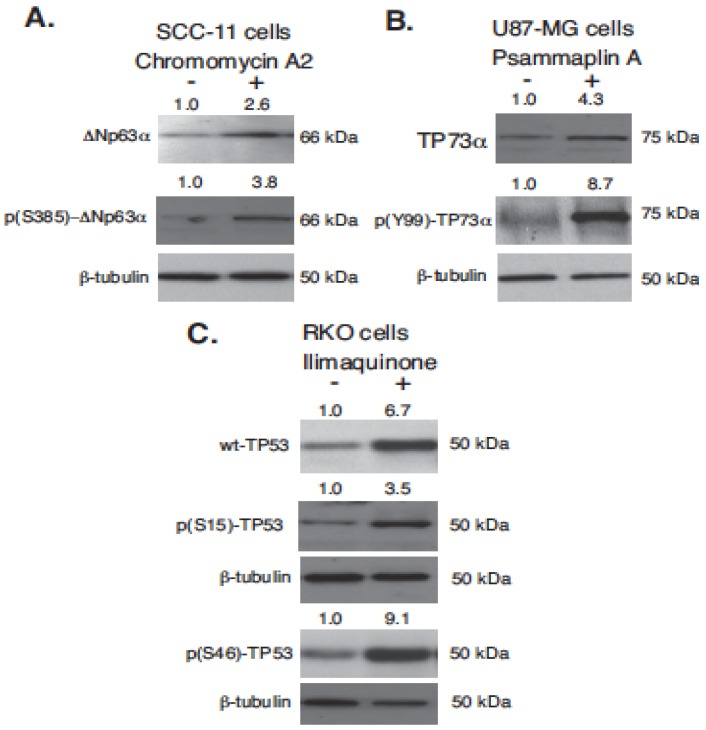
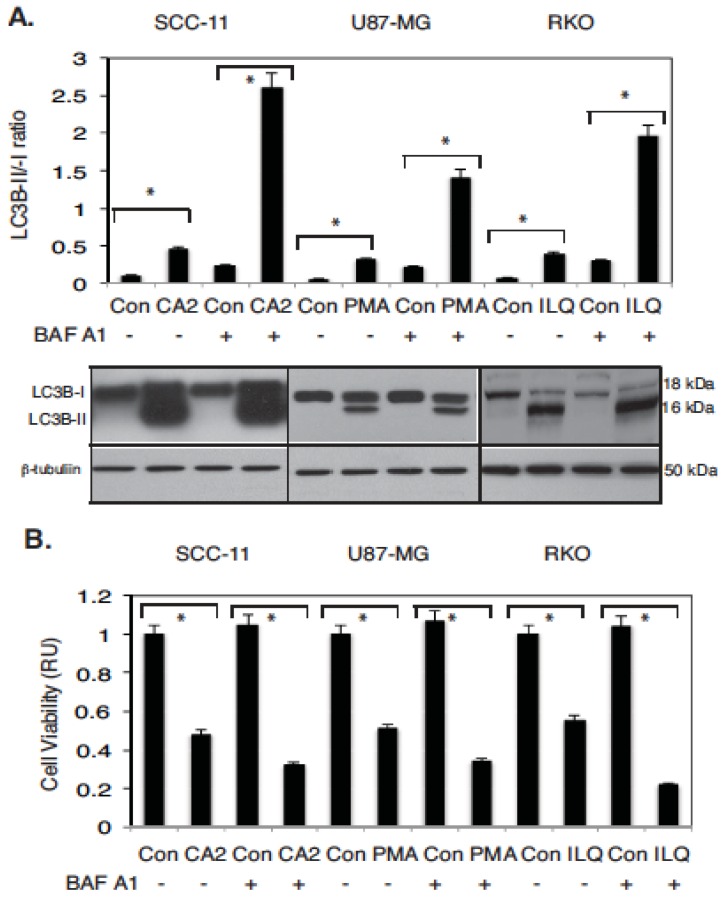
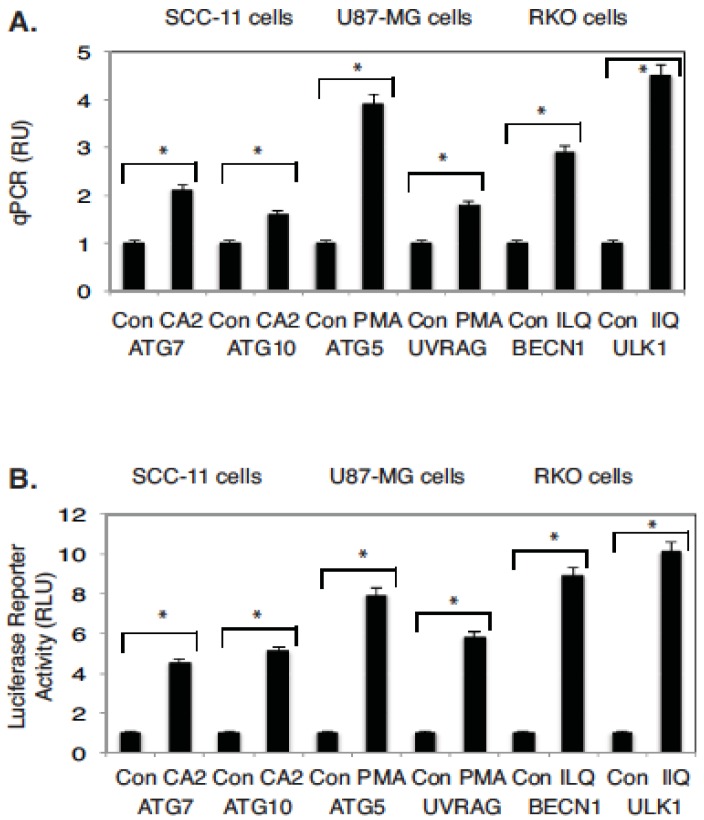
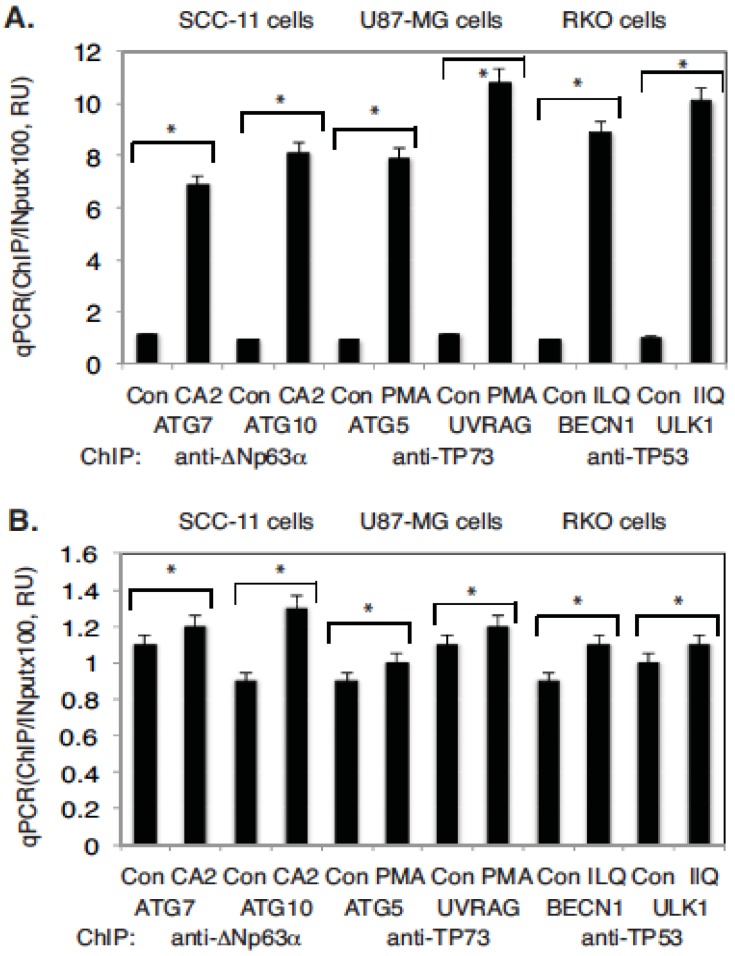
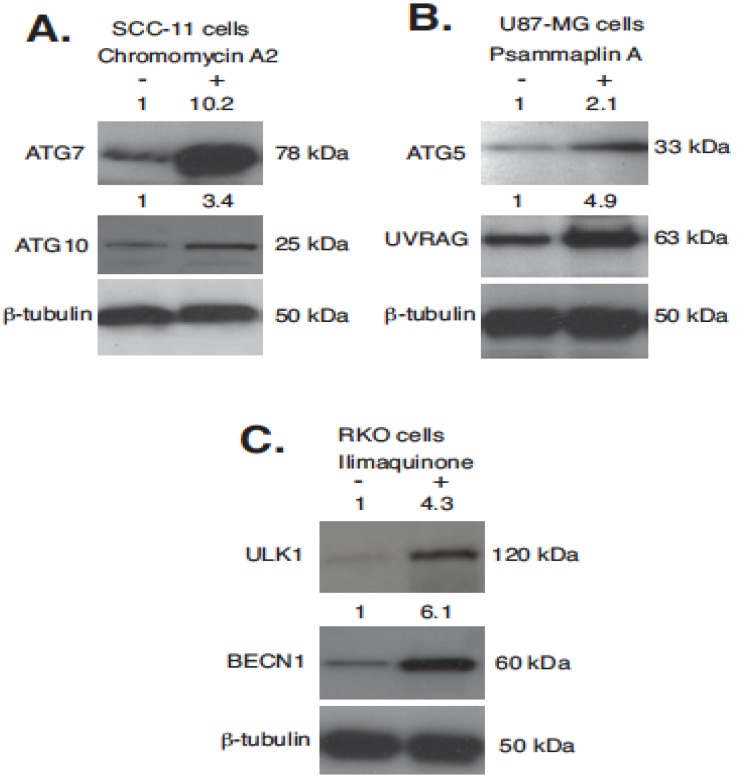
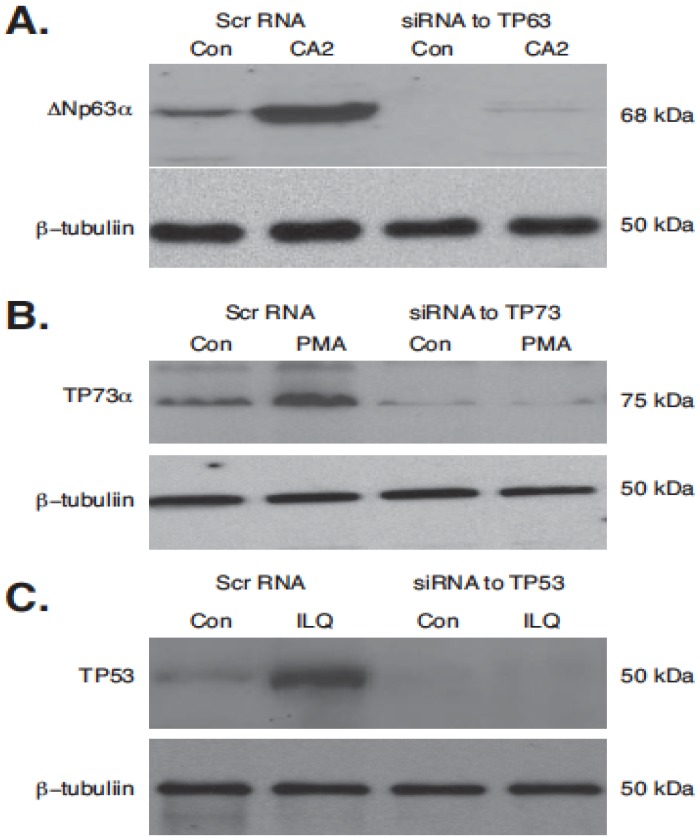
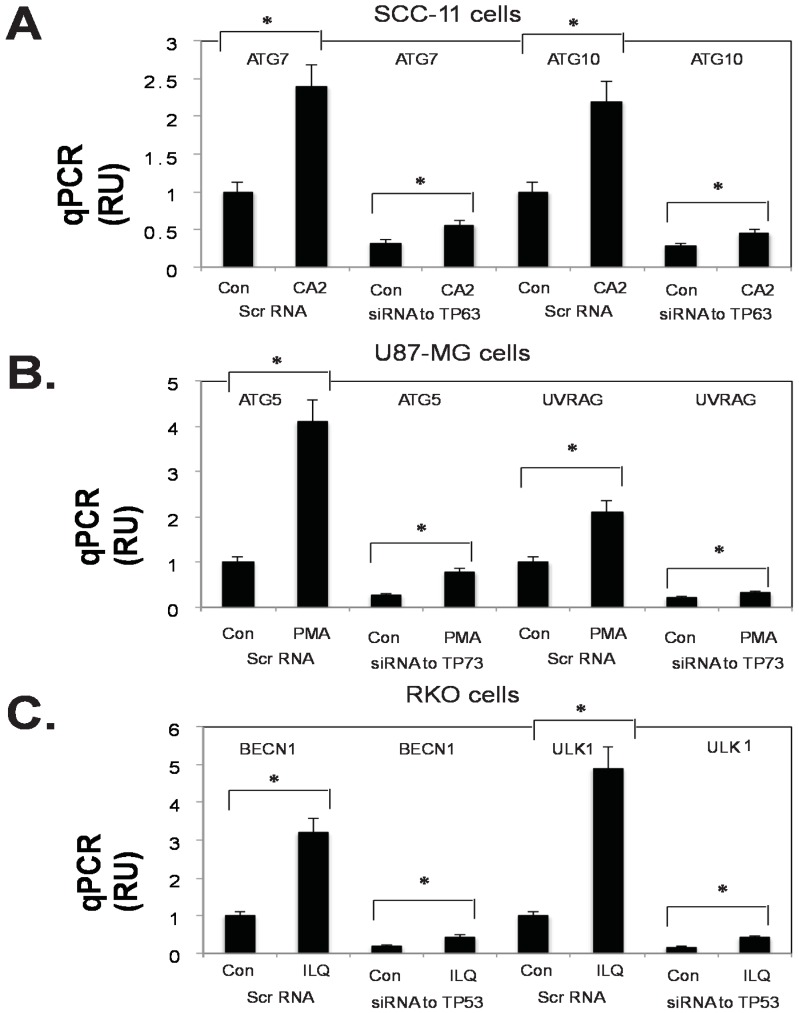
Targeting autophagic pathways might play a critical role in designing novel chemotherapeutic approaches in the treatment of human cancers, and the prevention of tumor-derived chemoresistance. Marine compounds were found to decrease tumor cell growth in vitro and in vivo. Some of them were shown to induce autophagic flux in tumor cells. In this study, we observed that the selected marine life-derived compounds (Chromomycin A2, Psammaplin A, and Ilimaquinone) induce expression of several autophagic signaling intermediates in human squamous cell carcinoma, glioblastoma, and colorectal carcinoma cells in vitro through a transcriptional regulation by tumor protein (TP)-p53 family members. These conclusions were supported by specific qPCR expression analysis, luciferase reporter promoter assay, and chromatin immunoprecipitation of promoter sequences bound to the TP53 family proteins, and silencing of the TP53 members in tumor cells.
Keywords: autophagy; cancer; marine drugs; p53 family members; transcription.
Figures
Similar articles
-
Marine-Derived Natural Lead Compound Disulfide-Linked Dimer Psammaplin A: Biological Activity and Structural Modification.Mar Drugs. 2019 Jun 27;17(7):384. doi: 10.3390/md17070384. Mar Drugs. 2019. PMID: 31252563 Free PMC article. Review.
-
Activation of p53 with ilimaquinone and ethylsmenoquinone, marine sponge metabolites, induces apoptosis and autophagy in colon cancer cells.Mar Drugs. 2015 Jan 16;13(1):543-57. doi: 10.3390/md13010543. Mar Drugs. 2015. PMID: 25603347 Free PMC article.
-
Imiquimod activates p53-dependent apoptosis in a human basal cell carcinoma cell line.J Dermatol Sci. 2016 Mar;81(3):182-91. doi: 10.1016/j.jdermsci.2015.12.011. Epub 2015 Dec 29. J Dermatol Sci. 2016. PMID: 26775629
-
Psammaplin A induces Sirtuin 1-dependent autophagic cell death in doxorubicin-resistant MCF-7/adr human breast cancer cells and xenografts.Biochim Biophys Acta. 2015 Feb;1850(2):401-10. doi: 10.1016/j.bbagen.2014.11.007. Epub 2014 Nov 12. Biochim Biophys Acta. 2015. PMID: 25445714
-
Apoptosis inducing lead compounds isolated from marine organisms of potential relevance in cancer treatment.Mutat Res. 2014 Oct;768:84-97. doi: 10.1016/j.mrfmmm.2014.03.005. Epub 2014 Mar 28. Mutat Res. 2014. PMID: 24685981 Review.
Cited by
-
Marine-Derived Natural Lead Compound Disulfide-Linked Dimer Psammaplin A: Biological Activity and Structural Modification.Mar Drugs. 2019 Jun 27;17(7):384. doi: 10.3390/md17070384. Mar Drugs. 2019. PMID: 31252563 Free PMC article. Review.
-
Mechanism of irreversible inhibition of Mycobacterium tuberculosis shikimate kinase by ilimaquinone.Biochim Biophys Acta Proteins Proteom. 2018 May-Jun;1866(5-6):731-739. doi: 10.1016/j.bbapap.2018.04.007. Epub 2018 Apr 12. Biochim Biophys Acta Proteins Proteom. 2018. PMID: 29654976 Free PMC article.
-
Natural Products as Modulators of the Proteostasis Machinery: Implications in Neurodegenerative Diseases.Int J Mol Sci. 2019 Sep 20;20(19):4666. doi: 10.3390/ijms20194666. Int J Mol Sci. 2019. PMID: 31547084 Free PMC article. Review.
-
Marine Compounds and Autophagy: Beginning of a New Era.Mar Drugs. 2018 Jul 31;16(8):260. doi: 10.3390/md16080260. Mar Drugs. 2018. PMID: 30065190 Free PMC article. No abstract available.
-
Marine Invertebrates: A Promissory Still Unexplored Source of Inhibitors of Biomedically Relevant Metallo Aminopeptidases Belonging to the M1 and M17 Families.Mar Drugs. 2023 Apr 28;21(5):279. doi: 10.3390/md21050279. Mar Drugs. 2023. PMID: 37233473 Free PMC article. Review.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
