

Clinical course of 63 patients with neonatal onset urea cycle disorders in the years 2001-2013
- PMID: 27538463
- PMCID: PMC4991093
- DOI: 10.1186/s13023-016-0493-0
Clinical course of 63 patients with neonatal onset urea cycle disorders in the years 2001-2013
Abstract
Background: Urea cycle disorders (UCDs) are rare inherited metabolic defects of ammonia detoxification. In about half of patients presenting with a UCD, the first symptoms appear within a few days after birth. These neonatal onset patients generally have a severe defect of urea cycle function and their survival and outcome prognoses are often limited. To understand better the current situation of neonatal onset in UCDs, we have performed a multicentre, retrospective, non-interventional case series study focussing on the most severe UCDs, namely defects of carbamoyl phosphate synthetase 1 (CPS1), ornithine transcarbamylase (OTC), and argininosuccinate synthetase (ASS).
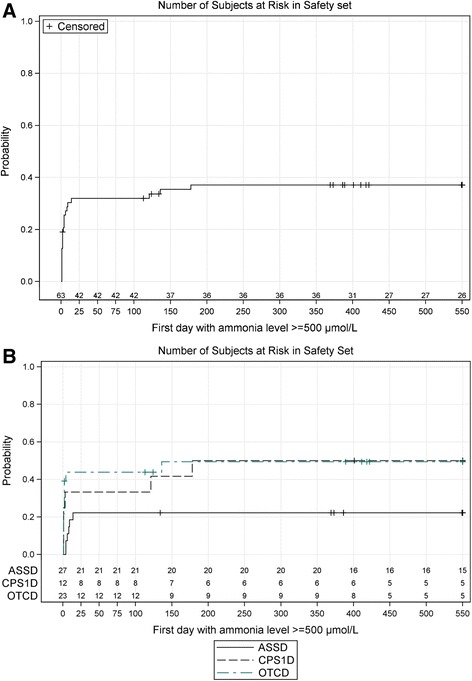
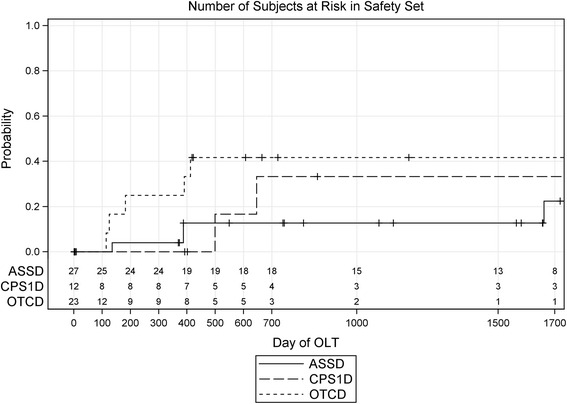
Methods and results: Data of 63 patients were collected (27 patients with ASS deficiency, 23 patients with OTC deficiency, and 12 patients with CPS1 deficiency, one patient definite diagnosis not documented). The majority of patients (43/63, 68 %) had an initial ammonia concentration exceeding 500 μmol/L (normal < 100), of which most (26/43, 60.5 %) were also encephalopathic and were treated with hemodialysis. In patients surviving the initial crisis, recurrence of hyperammonemic events within the first 1.5 years of life occurred frequently (mean 3.6 events, range 0-20). Of all patients, 16 (25.4 %) died during or immediately after the neonatal period.
Conclusion: We observed in this cohort of neonatal onset UCD patients a high rate of initial life-threatening hyperammonemia and a high risk of recurrence of severe hyperammonemic crises. These corresponded to a high mortality rate during the entire study period (30.2 %) despite the fact that patients were treated in leading European metabolic centers. This underlines the need to critically re-evaluate the current treatment strategies in these patients.
Keywords: Argininosuccinate synthetase deficiency; Carbamoyl phosphate synthetase 1 deficiency; Dialysis; Ornithine transcarbamylase deficiency; Urea cycle disorders; hyperammonemic crisis; neonatal hyperammonemia.
Figures
References
-
- Sokoro AA, Lepage J, Antonishyn N, McDonald R, Rockman-Greenberg C, Irvine J, Lehotay DC. Diagnosis and high incidence of hyperornithinemia-hyperammonemia-homocitrullinemia (HHH) syndrome in northern Saskatchewan. J Inherit Metab Dis. 2010;33(Suppl 3):S275–81. doi: 10.1007/s10545-010-9148-9. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
