

Novel Homozygous Missense Mutation in SPG20 Gene Results in Troyer Syndrome Associated with Mitochondrial Cytochrome c Oxidase Deficiency
- PMID: 27539578
- PMCID: PMC5413448
- DOI: 10.1007/8904_2016_580
Novel Homozygous Missense Mutation in SPG20 Gene Results in Troyer Syndrome Associated with Mitochondrial Cytochrome c Oxidase Deficiency
Abstract
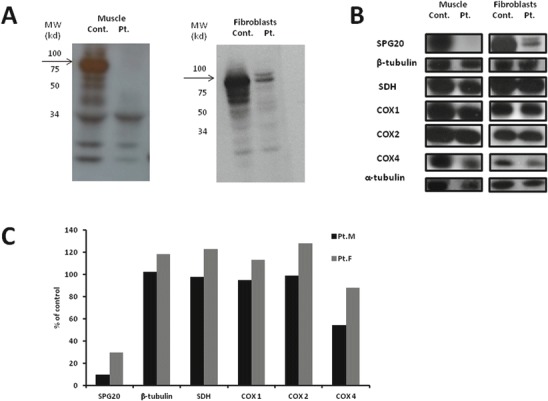
Troyer syndrome is an autosomal recessive form of hereditary spastic paraplegia (HSP) caused by deleterious mutations in the SPG20 gene. Although the disease is associated with a loss of function mechanism of spartin, the protein encoded by SPG20, the precise pathogenesis is yet to be elucidated. Recent data indicated an important role for spartin in both mitochondrial maintenance and function. Here we report a child presenting with progressive spastic paraparesis, generalized muscle weakness, dysarthria, impaired growth, and severe isolated decrease in muscle cytochrome c oxidase (COX) activity. Whole exome sequencing identified the homozygous c.988A>G variant in SPG20 gene (p.Met330Val) resulting in almost complete loss of spartin in skeletal muscle. Further analyses demonstrated significant tissue specific reduction of COX 4, a nuclear encoded subunit of COX, in muscle suggesting a role for spartin in proper mitochondrial respiratory chain function mediated by COX activity. Our findings need to be verified in other Troyer syndrome patients in order to classify it as a form of HSP caused by mitochondrial dysfunction.
Keywords: Cytochrome c oxidase; Hereditary spastic paraplegia; Mitochondria; Oxidative phosphorylation; SPG20; Troyer syndrome.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
