

Integration of omic networks in a developmental atlas of maize
- PMID: 27540173
- PMCID: PMC5808982
- DOI: 10.1126/science.aag1125
Integration of omic networks in a developmental atlas of maize
Abstract
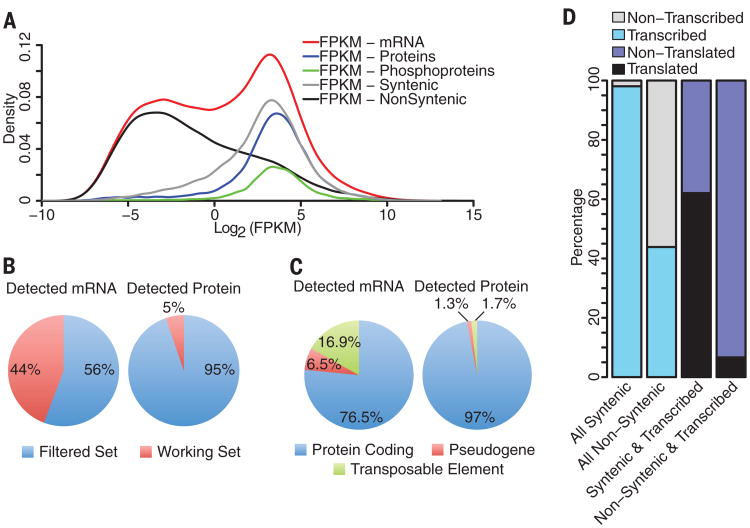
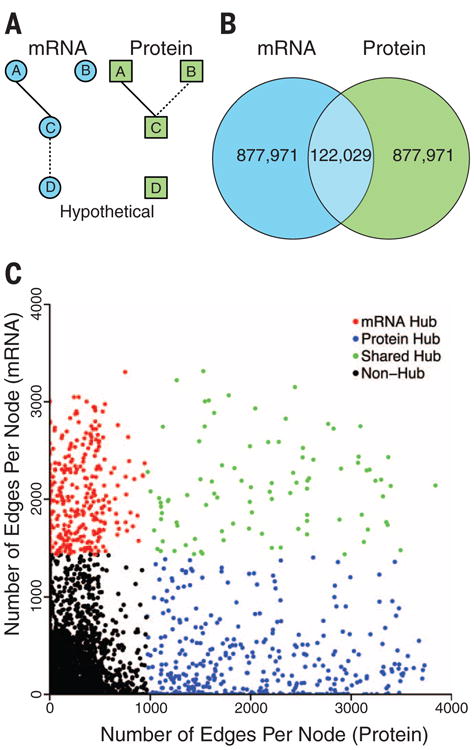
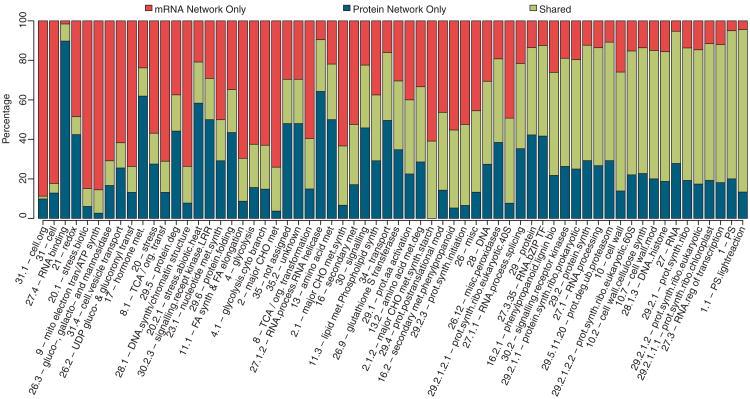
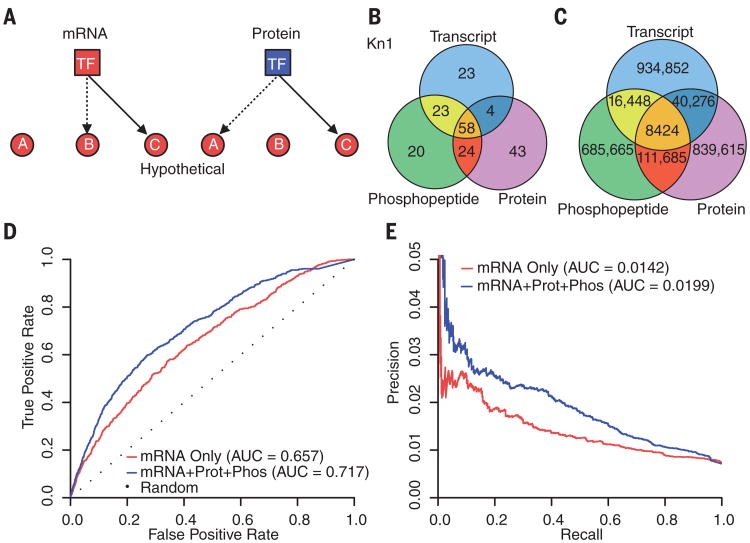
Coexpression networks and gene regulatory networks (GRNs) are emerging as important tools for predicting functional roles of individual genes at a system-wide scale. To enable network reconstructions, we built a large-scale gene expression atlas composed of 62,547 messenger RNAs (mRNAs), 17,862 nonmodified proteins, and 6227 phosphoproteins harboring 31,595 phosphorylation sites quantified across maize development. Networks in which nodes are genes connected on the basis of highly correlated expression patterns of mRNAs were very different from networks that were based on coexpression of proteins. Roughly 85% of highly interconnected hubs were not conserved in expression between RNA and protein networks. However, networks from either data type were enriched in similar ontological categories and were effective in predicting known regulatory relationships. Integration of mRNA, protein, and phosphoprotein data sets greatly improved the predictive power of GRNs.
Copyright © 2016, American Association for the Advancement of Science.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
