

Why Cockayne syndrome patients do not get cancer despite their DNA repair deficiency
- PMID: 27543334
- PMCID: PMC5018765
- DOI: 10.1073/pnas.1610020113
Why Cockayne syndrome patients do not get cancer despite their DNA repair deficiency
Abstract
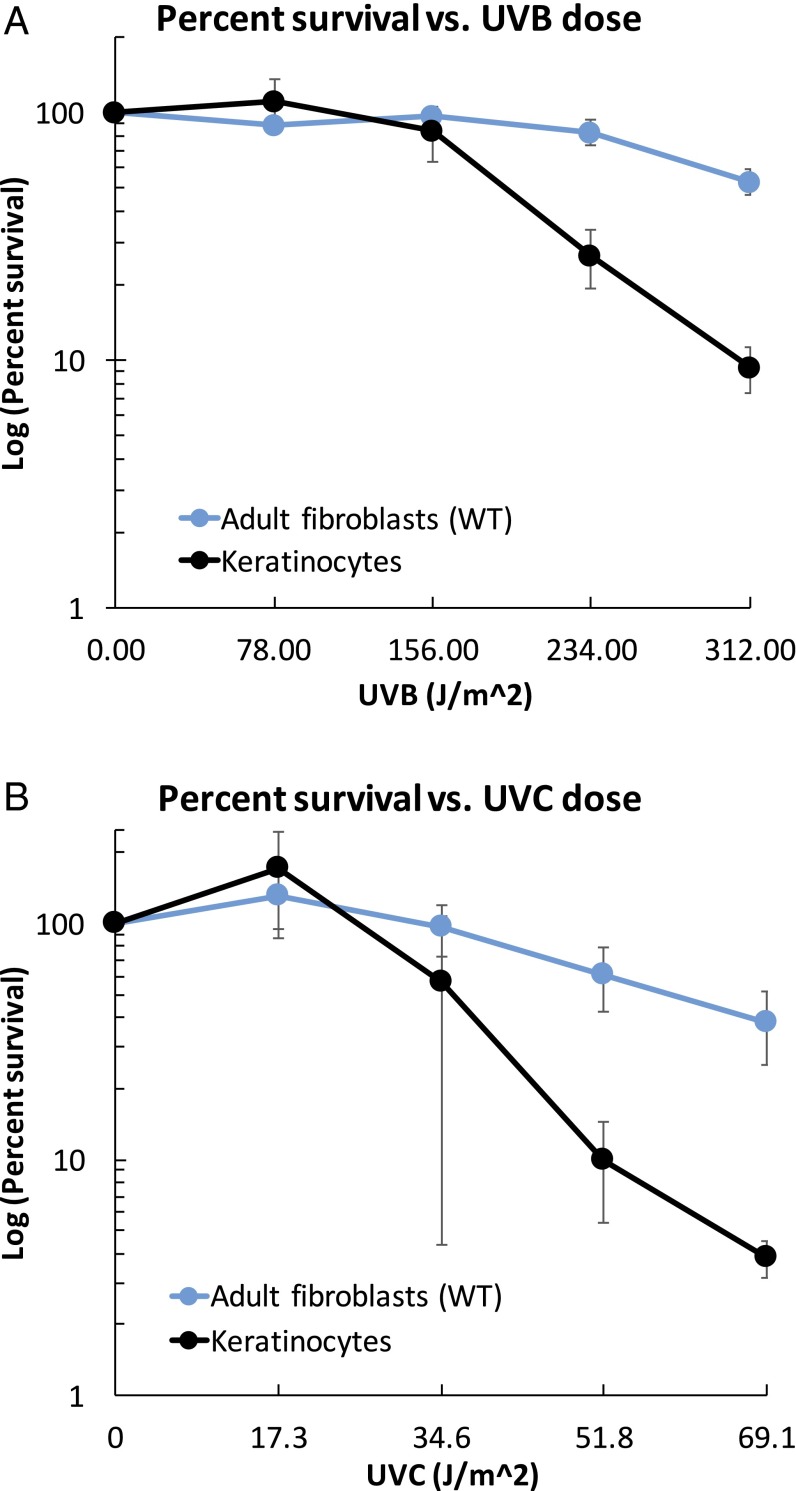
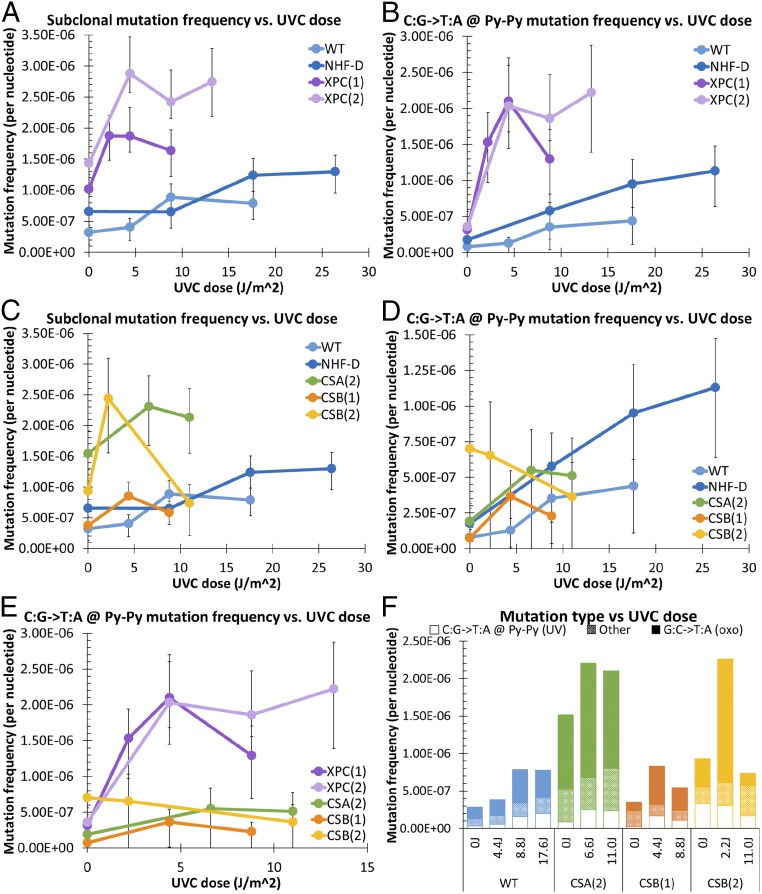
Cockayne syndrome (CS) and xeroderma pigmentosum (XP) are human photosensitive diseases with mutations in the nucleotide excision repair (NER) pathway, which repairs DNA damage from UV exposure. CS is mutated in the transcription-coupled repair (TCR) branch of the NER pathway and exhibits developmental and neurological pathologies. The XP-C group of XP patients have mutations in the global genome repair (GGR) branch of the NER pathway and have a very high incidence of UV-induced skin cancer. Cultured cells from both diseases have similar sensitivity to UV-induced cytotoxicity, but CS patients have never been reported to develop cancer, although they often exhibit photosensitivity. Because cancers are associated with increased mutations, especially when initiated by DNA damage, we examined UV-induced mutagenesis in both XP-C and CS cells, using duplex sequencing for high-sensitivity mutation detection. Duplex sequencing detects rare mutagenic events, independent of selection and in multiple loci, enabling examination of all mutations rather than just those that confer major changes to a specific protein. We found telomerase-positive normal and CS-B cells had increased background mutation frequencies that decreased upon irradiation, purging the population of subclonal variants. Primary XP-C cells had increased UV-induced mutation frequencies compared with normal cells, consistent with their GGR deficiency. CS cells, in contrast, had normal levels of mutagenesis despite their TCR deficiency. The lack of elevated UV-induced mutagenesis in CS cells reveals that their TCR deficiency, although increasing cytotoxicity, is not mutagenic. Therefore the absence of cancer in CS patients results from the absence of UV-induced mutagenesis rather than from enhanced lethality.
Keywords: RNA pol II; apoptosis; dipyrimidines; mutagenesis; transcription arrest.
Conflict of interest statement
The authors declare no conflict of interest.
Figures







Similar articles
-
DNA repair and ultraviolet mutagenesis in cells from a new patient with xeroderma pigmentosum group G and cockayne syndrome resemble xeroderma pigmentosum cells.J Invest Dermatol. 1996 Oct;107(4):647-53. doi: 10.1111/1523-1747.ep12584287. J Invest Dermatol. 1996. PMID: 8823375
-
UV-enhanced reactivation of a UV-damaged reporter gene suggests transcription-coupled repair is UV-inducible in human cells.Carcinogenesis. 1999 Jan;20(1):19-26. doi: 10.1093/carcin/20.1.19. Carcinogenesis. 1999. PMID: 9934845
-
Sources and consequences of oxidative damage from mitochondria and neurotransmitter signaling.Environ Mol Mutagen. 2016 Jun;57(5):322-30. doi: 10.1002/em.21995. Epub 2016 Jan 14. Environ Mol Mutagen. 2016. PMID: 27311994 Review.
-
Sirt1 suppresses RNA synthesis after UV irradiation in combined xeroderma pigmentosum group D/Cockayne syndrome (XP-D/CS) cells.Proc Natl Acad Sci U S A. 2013 Jan 15;110(3):E212-20. doi: 10.1073/pnas.1213076110. Epub 2012 Dec 24. Proc Natl Acad Sci U S A. 2013. PMID: 23267107 Free PMC article.
-
Common pathways for ultraviolet skin carcinogenesis in the repair and replication defective groups of xeroderma pigmentosum.J Dermatol Sci. 2000 May;23(1):1-11. doi: 10.1016/s0923-1811(99)00088-2. J Dermatol Sci. 2000. PMID: 10699759 Review.
Cited by
-
Genomic mutation landscape of skin cancers from DNA repair-deficient xeroderma pigmentosum patients.Nat Commun. 2023 May 4;14(1):2561. doi: 10.1038/s41467-023-38311-0. Nat Commun. 2023. PMID: 37142601 Free PMC article.
-
DNA damage and mitochondria in cancer and aging.Carcinogenesis. 2020 Dec 31;41(12):1625-1634. doi: 10.1093/carcin/bgaa114. Carcinogenesis. 2020. PMID: 33146705 Free PMC article. Review.
-
Aging and cancer.Mol Cancer. 2024 May 18;23(1):106. doi: 10.1186/s12943-024-02020-z. Mol Cancer. 2024. PMID: 38760832 Free PMC article. Review.
-
Cockayne Syndrome Group B (CSB): The Regulatory Framework Governing the Multifunctional Protein and Its Plausible Role in Cancer.Cells. 2021 Apr 10;10(4):866. doi: 10.3390/cells10040866. Cells. 2021. PMID: 33920220 Free PMC article. Review.
-
Drugging the Cancers Addicted to DNA Repair.J Natl Cancer Inst. 2017 Nov 1;109(11):djx059. doi: 10.1093/jnci/djx059. J Natl Cancer Inst. 2017. PMID: 28521333 Free PMC article. Review.
References
-
- Cleaver JE, Lam ET, Revet I. Disorders of nucleotide excision repair: The genetic and molecular basis of heterogeneity. Nat Rev Genet. 2009;10(11):756–768. - PubMed
-
- Kraemer KH, Lee MM, Scotto J. DNA repair protects against cutaneous and internal neoplasia: Evidence from xeroderma pigmentosum. Carcinogenesis. 1984;5(4):511–514. - PubMed
-
- Nance MA, Berry SA. Cockayne syndrome: Review of 140 cases. Am J Med Genet. 1992;42(1):68–84. - PubMed
-
- Zhang WR, Garrett GL, Cleaver JE, Arron ST. Absence of skin cancer in the DNA repair-deficient disease Cockayne Syndrome (CS): A survey study. J Am Acad Dermatol. 2016;74(6):1270–1272. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
