

How cryo-electron microscopy and X-ray crystallography complement each other
- PMID: 27543495
- PMCID: PMC5192981
- DOI: 10.1002/pro.3022
How cryo-electron microscopy and X-ray crystallography complement each other
Abstract
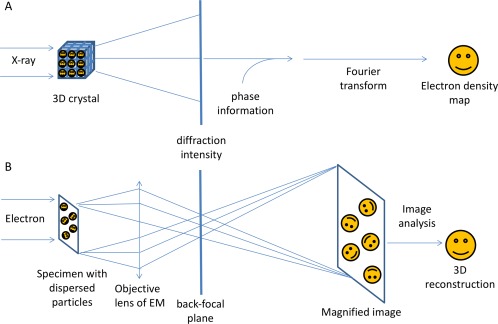
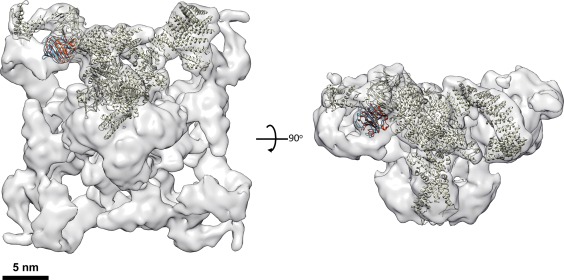
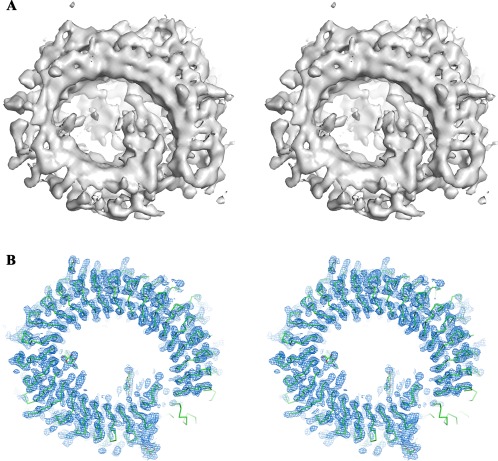
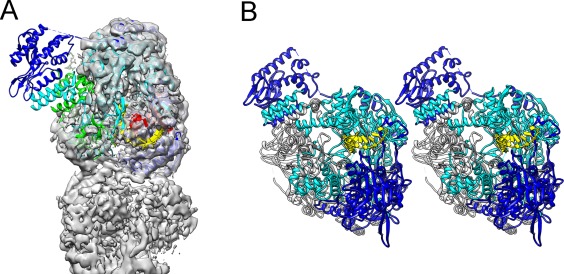
With the ability to resolve structures of macromolecules at atomic resolution, X-ray crystallography has been the most powerful tool in modern structural biology. At the same time, recent technical improvements have triggered a resolution revolution in the single particle cryo-EM method. While the two methods are different in many respects, from sample preparation to structure determination, they both have the power to solve macromolecular structures at atomic resolution. It is important to understand the unique advantages and caveats of the two methods in solving structures and to appreciate the complementary nature of the two methods in structural biology. In this review we provide some examples, and discuss how X-ray crystallography and cryo-EM can be combined in deciphering structures of macromolecules for our full understanding of their biological mechanisms.
Keywords: X-ray and electron scattering; X-ray crystallography; cryo-EM; protein structure determination; structural biology; structure determination methods.
© 2016 The Protein Society.
Figures
References
-
- Shi Y (2014) A glimpse of structural biology through X‐ray crystallography. Cell 159:995–1014. - PubMed
-
- Kuhlbrandt W (2014) Biochemistry. The resolution revolution. Science 343:1443–1444. - PubMed
-
- Newman J (2006) A review of techniques for maximizing diffraction from a protein crystal in stilla. Acta Cryst D62:27–31. - PubMed
-
- Rossmann MG (2000) Fitting atomic models into electron‐microscopy maps. Acta Cryst D56:1341–1349. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
