

Inherited epidermolysis bullosa and squamous cell carcinoma: a systematic review of 117 cases
- PMID: 27544590
- PMCID: PMC4992553
- DOI: 10.1186/s13023-016-0489-9
Inherited epidermolysis bullosa and squamous cell carcinoma: a systematic review of 117 cases
Abstract
Background: Inherited epidermolysis bullosa (EB) comprises a highly heterogeneous group of rare diseases characterized by exacerbated skin and/or mucosal fragility and blister formation after minor mechanical trauma. Level of cleavage in the skin, clinical features with immunofluorescence antigen mapping and/or electron microscopy examination of a skin biopsy and/or gene involved, type(s) of mutation present and sometimes specific mutation(s), allow to define the EB type and subtype. This family of genodermatoses exposes patients to several complications, cutaneous squamous cell carcinoma (cSCC) being the most severe of them.
Objective: The aim of this systematic review was to document patients with EB who developed cSCC.
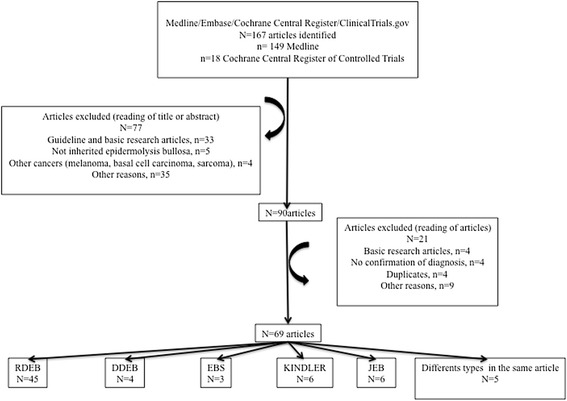
Methods: A systematic literature search was performed, from inception to March 2014, using Medline, Embase, Cochrane and ClinicalTrials.gov databases. Only articles published in English and French were selected. The diagnosis of EB had to be confirmed by EM and/or IFM and/or mutation analysis, while cSCC had to be confirmed by histological analysis.
Results: Of 167 references in the original search, 69 relevant articles were identified, representing 117 cases. cSCCs were identified in all types of EB, though predominantly in recessive dystrophic EB (RDEB) forms (81 cases (69.2 %)). The median age at diagnosis was 36 years old (interquartile range (IQR), 27-48 years and range, 6-71 years) for all forms. Of those with measurements in the literature (88 cases (75.2 %)), tumor size was greater than 2 centimeters in 52 cases (59.1 %). The histopathological characteristics were specified in 88 cases (75.2 %) and well-differentiated forms predominated (73.9 %). No conclusion could be drawn on the choice of surgical treatment or the management in advanced forms.
Limitations: This study was retrospective and statistical analysis was not included due to various biases. This study design did not allow to infer prevalence, nor EB subtype risk for cSCC occurrence.
Conclusions: Our study correlated with historical data shows that most of the cSCCs occurred in subjects with the RDEB subtype, however reports also show that cSCCs can present in any patients with EB. The first signs of cSCC developed at a younger age in EB patients than in non-EB patients. Interestingly, the cSCC duration, before its diagnosis, was shorter in individuals with RDEB than with junctional EB (JEB) and dominant dystrophic EB (DDEB). This study further emphasizes the importance of regular monitoring of EB patients, particularly with the RDEB subtype as they developed cSCC at a younger age.
Keywords: Chemotherapy; Cutaneous squamous cell carcinoma; Inherited epidermolysis bullosa; Radiotherapy; Systematic review; Target therapy.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
