

Cellular Cholesterol Directly Activates Smoothened in Hedgehog Signaling
- PMID: 27545348
- PMCID: PMC5035717
- DOI: 10.1016/j.cell.2016.08.003
Cellular Cholesterol Directly Activates Smoothened in Hedgehog Signaling
Abstract
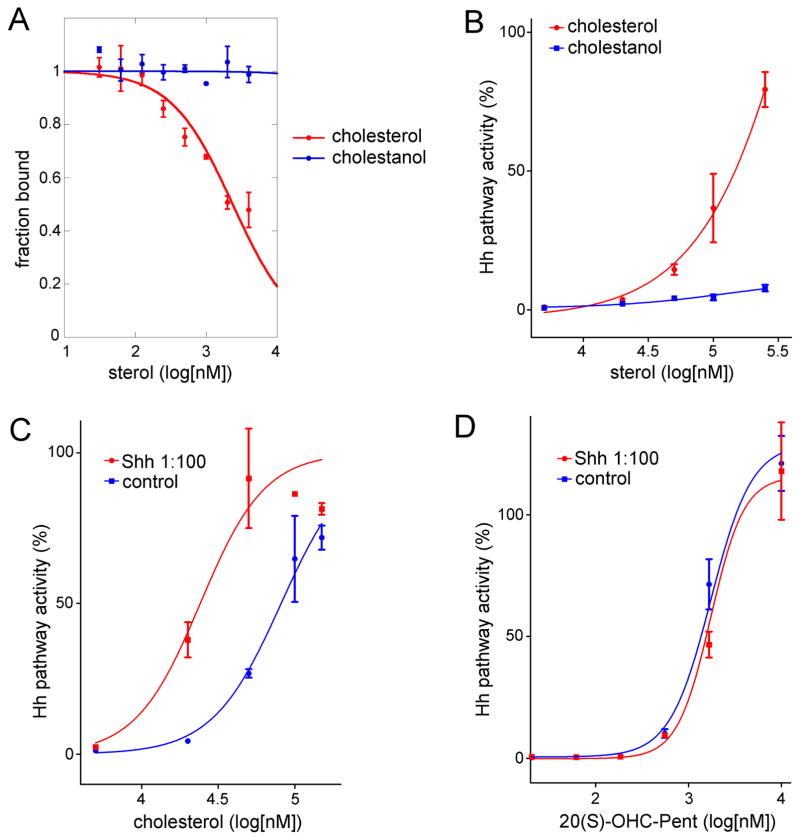
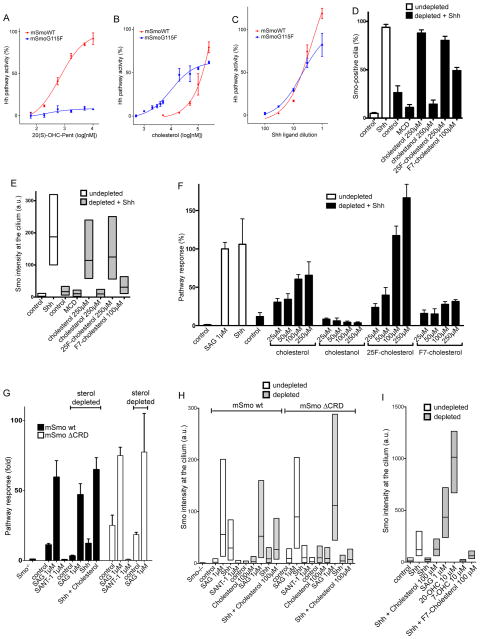
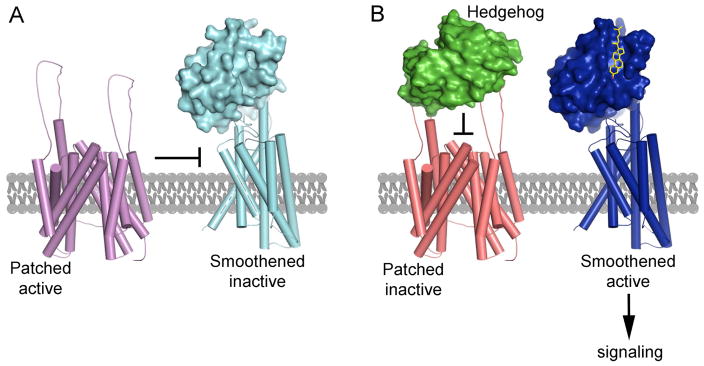
In vertebrates, sterols are necessary for Hedgehog signaling, a pathway critical in embryogenesis and cancer. Sterols activate the membrane protein Smoothened by binding its extracellular, cysteine-rich domain (CRD). Major unanswered questions concern the nature of the endogenous, activating sterol and the mechanism by which it regulates Smoothened. We report crystal structures of CRD complexed with sterols and alone, revealing that sterols induce a dramatic conformational change of the binding site, which is sufficient for Smoothened activation and is unique among CRD-containing receptors. We demonstrate that Hedgehog signaling requires sterol binding to Smoothened and define key residues for sterol recognition and activity. We also show that cholesterol itself binds and activates Smoothened. Furthermore, the effect of oxysterols is abolished in Smoothened mutants that retain activation by cholesterol and Hedgehog. We propose that the endogenous Smoothened activator is cholesterol, not oxysterols, and that vertebrate Hedgehog signaling controls Smoothened by regulating its access to cholesterol.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures
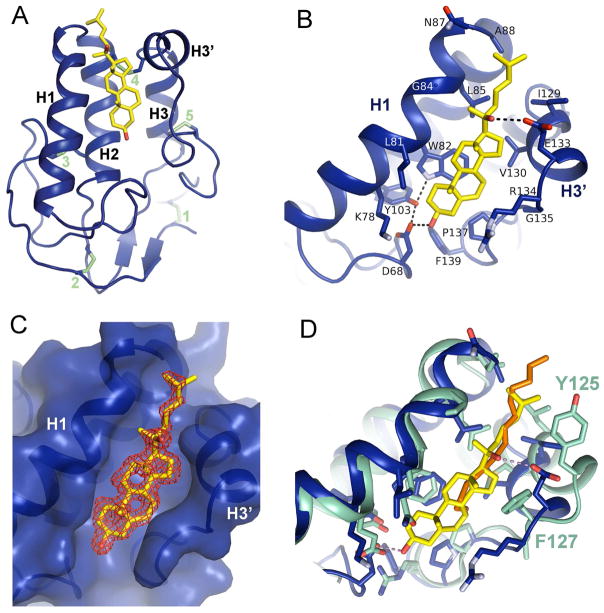
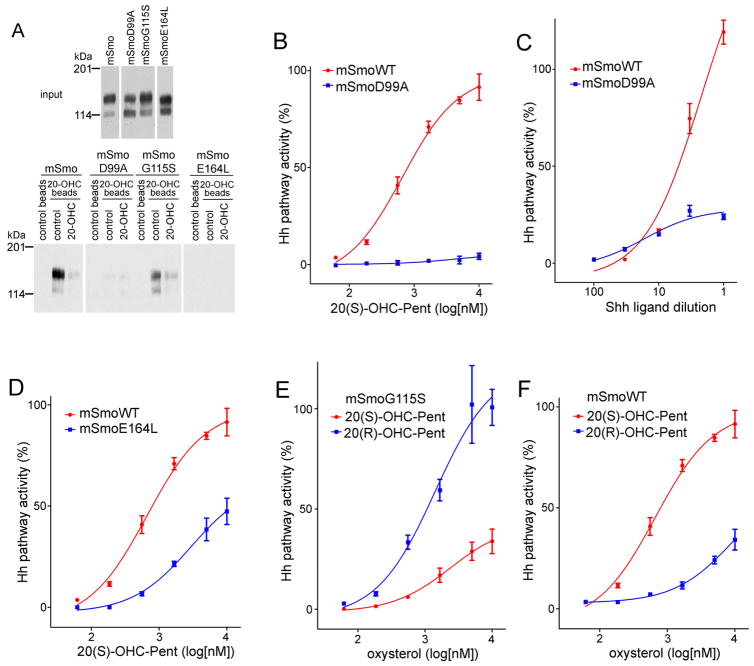
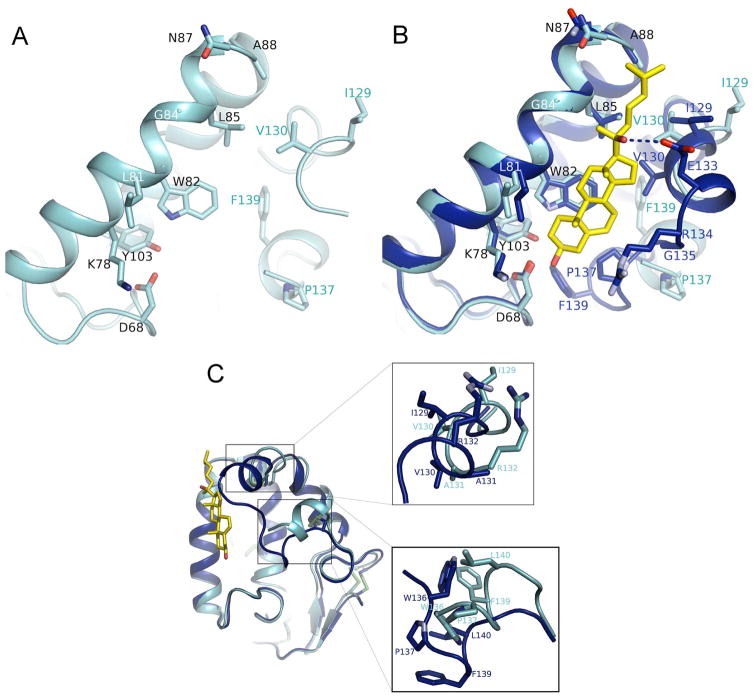
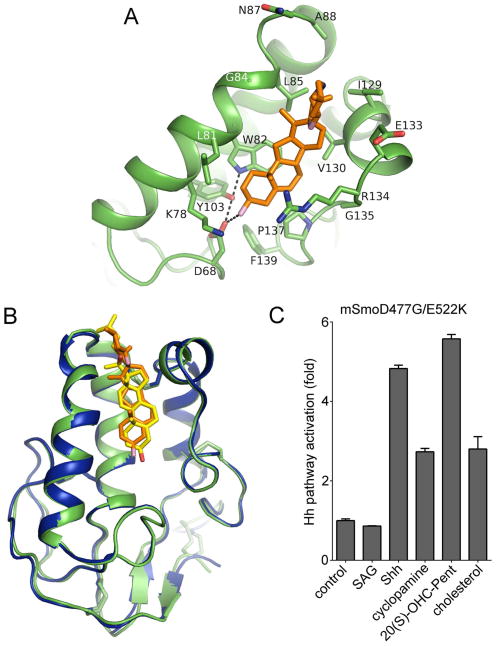
References
-
- Alcedo J, Ayzenzon M, Von Ohlen T, Noll M, Hooper JE. The Drosophila smoothened gene encodes a seven-pass membrane protein, a putative receptor for the hedgehog signal. Cell. 1996;86:221–232. - PubMed
-
- Bazan JF, de Sauvage FJ. Structural ties between cholesterol transport and morphogen signaling. Cell. 2009;138:1055–1056. - PubMed
-
- Carroll JN, Pinkerton FD, Su X, Gerst N, Wilson WK, Schroepfer GJ., Jr Sterol synthesis. Synthesis of 3 beta-hydroxy-25,26,26,26,27,27,27-heptafluorocholest-5-en-7-one and its effects on HMG-CoA reductase activity in Chinese hamster ovary cells, on ACAT activity in rat jejunal microsomes, and serum cholesterol levels in rats. Chemistry and physics of lipids. 1998;94:209–225. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
