

Determinants of Power in Gene-Based Burden Testing for Monogenic Disorders
- PMID: 27545677
- PMCID: PMC5011058
- DOI: 10.1016/j.ajhg.2016.06.031
Determinants of Power in Gene-Based Burden Testing for Monogenic Disorders
Abstract
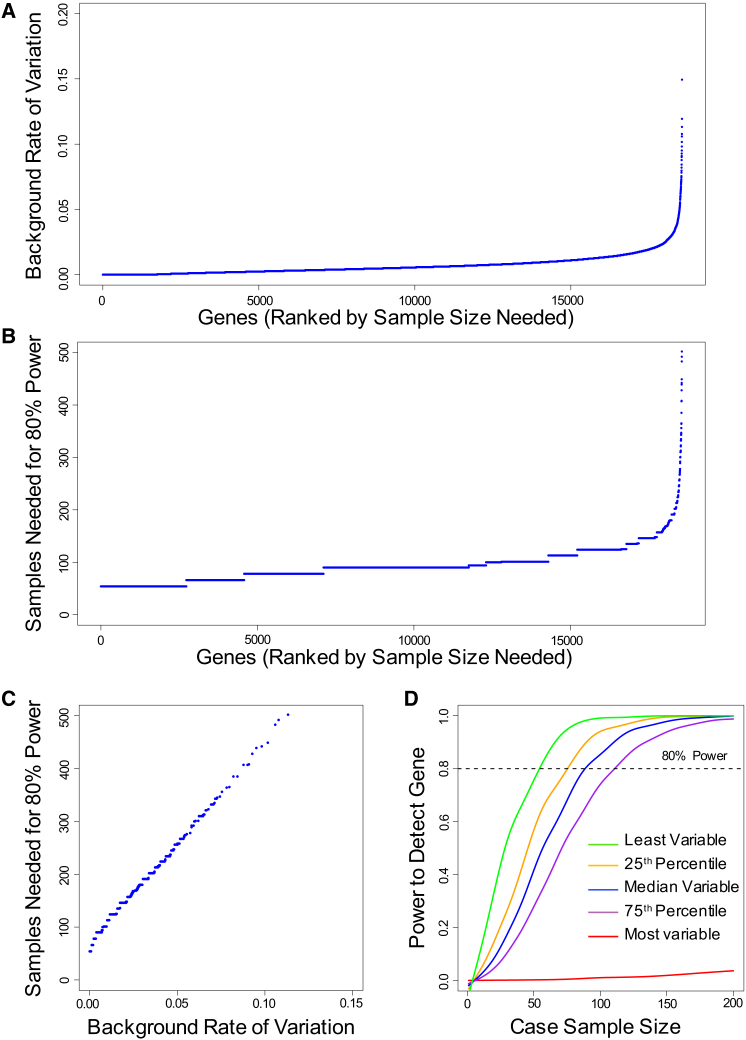
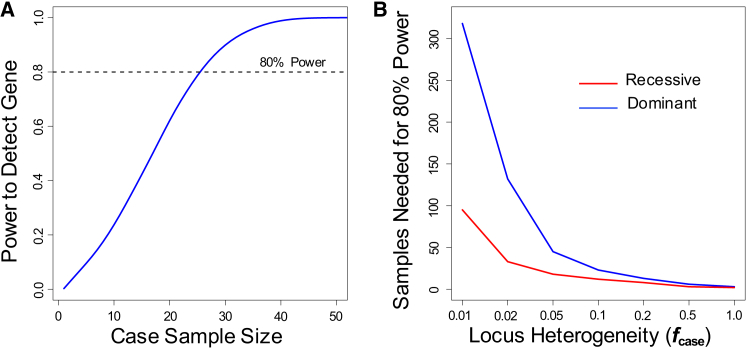
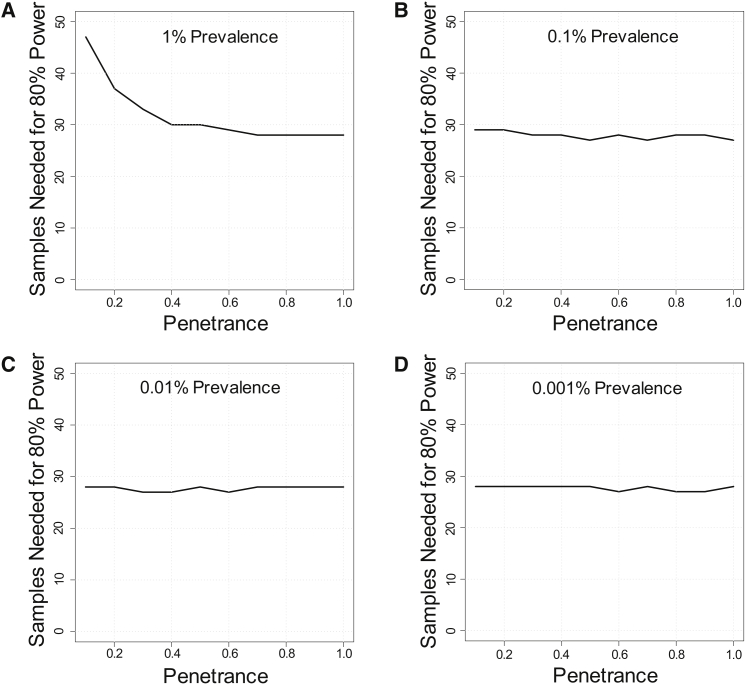
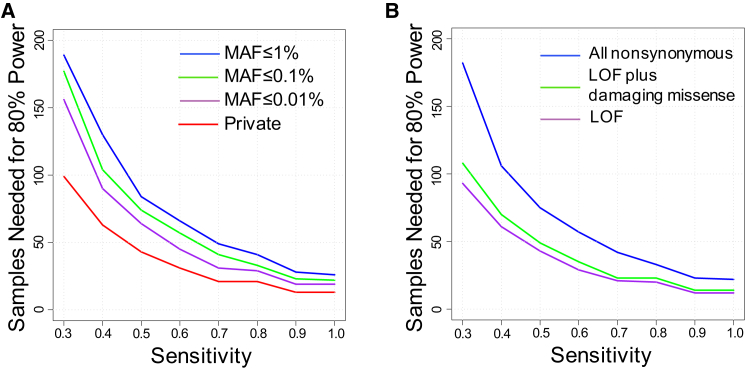
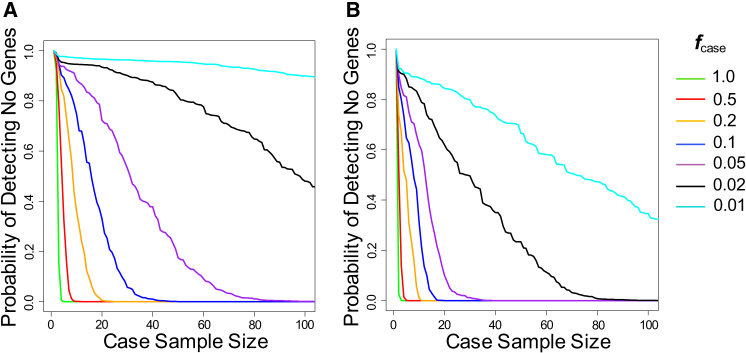
Whole-exome sequencing has enabled new approaches for discovering genes associated with monogenic disorders. One such approach is gene-based burden testing, in which the aggregate frequency of "qualifying variants" is compared between case and control subjects for each gene. Despite substantial successes of this approach, the genetic causes for many monogenic disorders remain unknown or only partially known. It is possible that particular genetic architectures lower rates of discovery, but the influence of these factors on power has not been rigorously evaluated. Here, we leverage large-scale exome-sequencing data to create an empirically based simulation framework to evaluate the impact of key parameters (background variation rates, locus heterogeneity, mode of inheritance, penetrance) on power in gene-based burden tests in the context of monogenic disorders. Our results demonstrate that across genes, there is a wide range in sample sizes needed to achieve power due to differences in the background rate of rare variants in each gene. Increasing locus heterogeneity results in rapid increases in sample sizes needed to achieve adequate power, particularly when individual genes contribute to less than 5% of cases under a dominant model. Interestingly, incomplete penetrance as low as 10% had little effect on power due to the low prevalence of monogenic disorders. Our results suggest that moderate incomplete penetrance is not an obstacle in this gene-based burden testing approach but that dominant disorders with high locus heterogeneity will require large sample sizes. Our simulations also provide guidance on sample size needs and inform study design under various genetic architectures.
Copyright © 2016 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Chong J.X., Buckingham K.J., Jhangiani S.N., Boehm C., Sobreira N., Smith J.D., Harrell T.M., McMillin M.J., Wiszniewski W., Gambin T., Centers for Mendelian Genomics The genetic basis of Mendelian phenotypes: discoveries, challenges, and opportunities. Am. J. Hum. Genet. 2015;97:199–215. - PMC - PubMed
-
- Risch N., Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1517. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
