

Culture Independent Genomic Comparisons Reveal Environmental Adaptations for Altiarchaeales
- PMID: 27547202
- PMCID: PMC4975002
- DOI: 10.3389/fmicb.2016.01221
Culture Independent Genomic Comparisons Reveal Environmental Adaptations for Altiarchaeales
Abstract
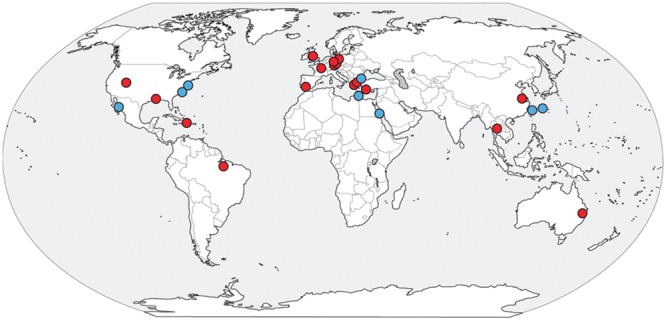
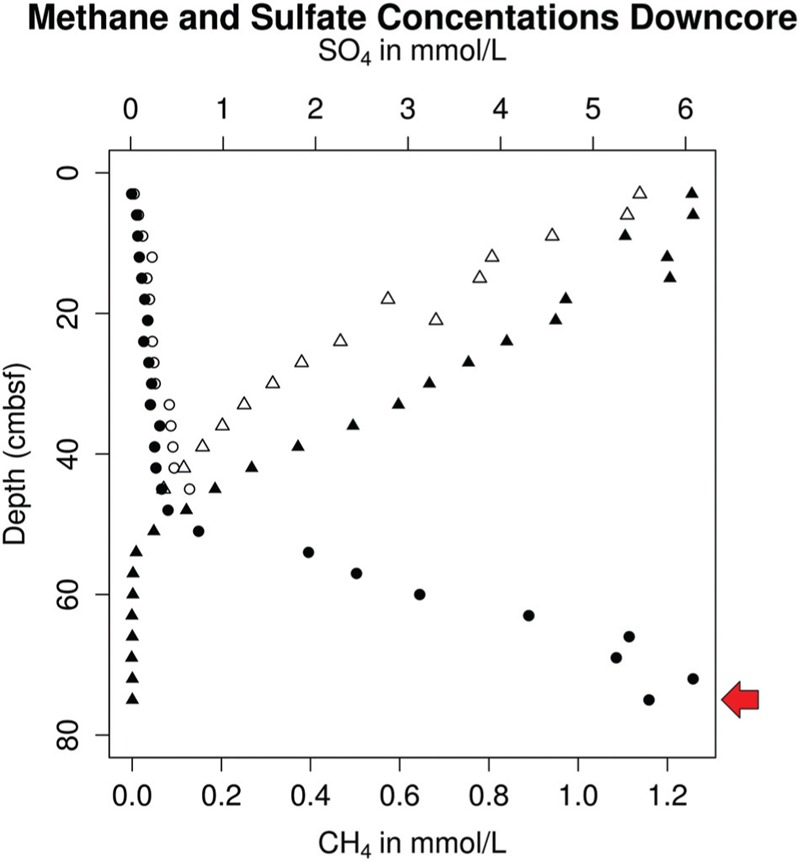
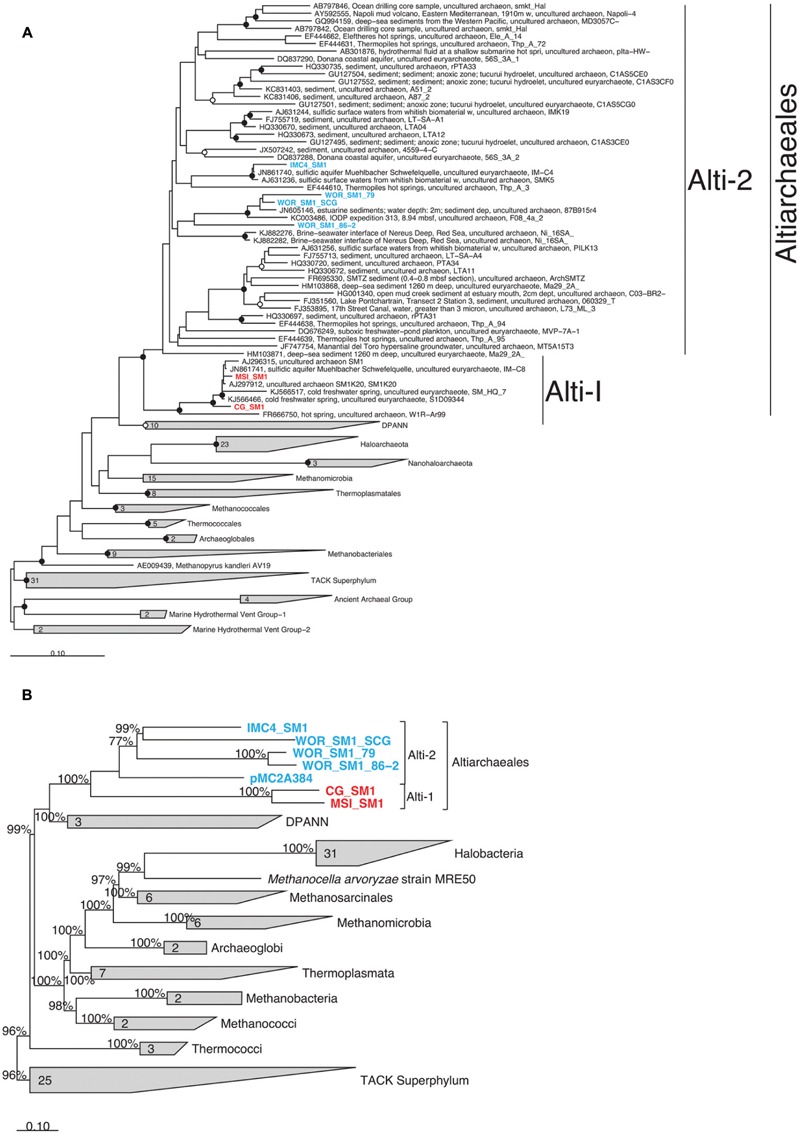
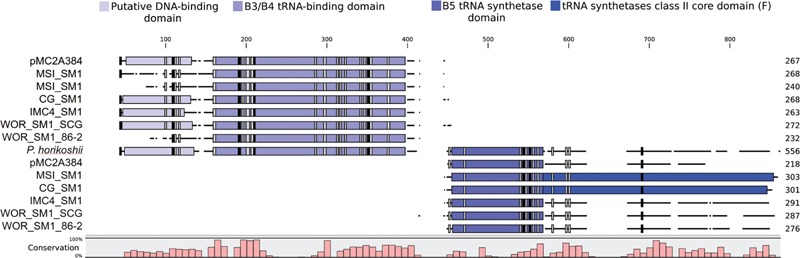
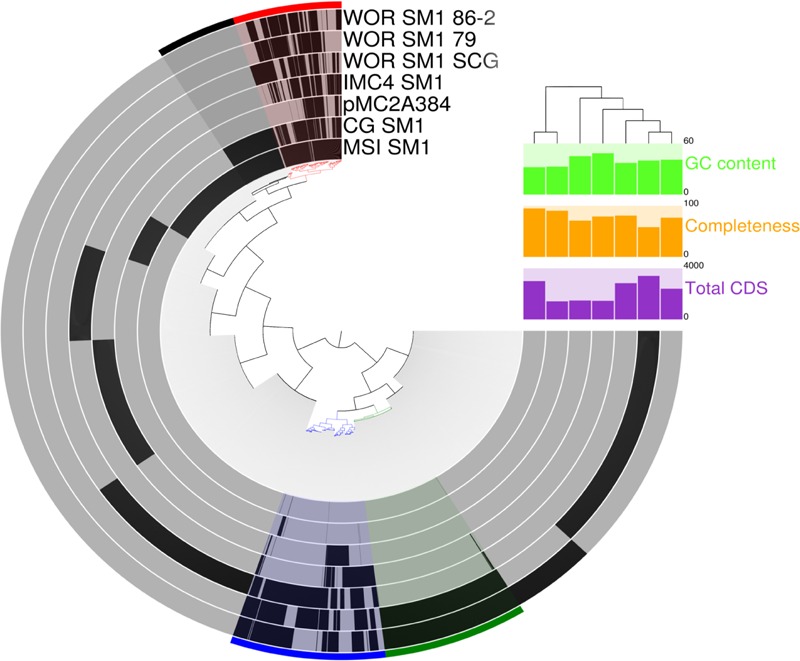
The recently proposed candidatus order Altiarchaeales remains an uncultured archaeal lineage composed of genetically diverse, globally widespread organisms frequently observed in anoxic subsurface environments. In spite of 15 years of studies on the psychrophilic biofilm-producing Candidatus Altiarchaeum hamiconexum and its close relatives, very little is known about the phylogenetic and functional diversity of the widespread free-living marine members of this taxon. From methanogenic sediments in the White Oak River Estuary, NC, USA, we sequenced a single cell amplified genome (SAG), WOR_SM1_SCG, and used it to identify and refine two high-quality genomes from metagenomes, WOR_SM1_79 and WOR_SM1_86-2, from the same site. These three genomic reconstructions form a monophyletic group, which also includes three previously published genomes from metagenomes from terrestrial springs and a SAG from Sakinaw Lake in a group previously designated as pMC2A384. A synapomorphic mutation in the Altiarchaeales tRNA synthetase β subunit, pheT, caused the protein to be encoded as two subunits at non-adjacent loci. Consistent with the terrestrial spring clades, our estuarine genomes contained a near-complete autotrophic metabolism, H2 or CO as potential electron donors, a reductive acetyl-CoA pathway for carbon fixation, and methylotroph-like NADP(H)-dependent dehydrogenase. Phylogenies based on 16S rRNA genes and concatenated conserved proteins identified two distinct sub-clades of Altiarchaeales, Alti-1 populated by organisms from actively flowing springs, and Alti-2 which was more widespread, diverse, and not associated with visible mats. The core Alti-1 genome suggested Alti-1 is adapted for the stream environment with lipopolysaccharide production capacity and extracellular hami structures. The core Alti-2 genome suggested members of this clade are free-living with distinct mechanisms for energy maintenance, motility, osmoregulation, and sulfur redox reactions. These data suggested that the hamus structures found in Candidatus Altiarchaeum hamiconexum are not present outside of stream-adapted Altiarchaeales. Homologs to a Na(+) transporter and membrane bound coenzyme A disulfide reductase that were unique to the brackish sediment Alti-2 genomes, could indicate adaptations to the estuarine, sulfur-rich environment.
Keywords: autotrophy; comparative genomics; ecophysiology; marine sediment; metagenomics; single cell genomics; uncultured archaea.
Figures
References
-
- Balestrino D., Ghigo J.-M., Charbonnel N., Haagensen J. A. J., Forestier C. (2008). The characterization of functions involved in the establishment and maturation of Klebsiella pneumoniae in vitro biofilm reveals dual roles for surface exopolysaccharides. Environ. Microbiol. 10 685–701. 10.1111/j.1462-2920.2007.01491.x - DOI - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases