

Genome-Wide SNP Calling from Genotyping by Sequencing (GBS) Data: A Comparison of Seven Pipelines and Two Sequencing Technologies
- PMID: 27547936
- PMCID: PMC4993469
- DOI: 10.1371/journal.pone.0161333
Genome-Wide SNP Calling from Genotyping by Sequencing (GBS) Data: A Comparison of Seven Pipelines and Two Sequencing Technologies
Abstract
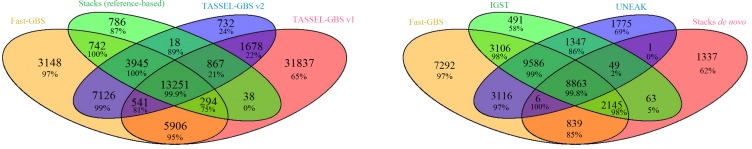
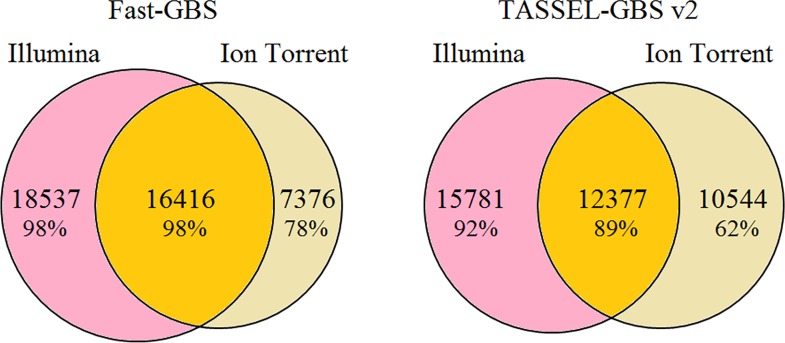
Next-generation sequencing (NGS) has revolutionized plant and animal research in many ways including new methods of high throughput genotyping. Genotyping-by-sequencing (GBS) has been demonstrated to be a robust and cost-effective genotyping method capable of producing thousands to millions of SNPs across a wide range of species. Undoubtedly, the greatest barrier to its broader use is the challenge of data analysis. Herein we describe a comprehensive comparison of seven GBS bioinformatics pipelines developed to process raw GBS sequence data into SNP genotypes. We compared five pipelines requiring a reference genome (TASSEL-GBS v1& v2, Stacks, IGST, and Fast-GBS) and two de novo pipelines that do not require a reference genome (UNEAK and Stacks). Using Illumina sequence data from a set of 24 re-sequenced soybean lines, we performed SNP calling with these pipelines and compared the GBS SNP calls with the re-sequencing data to assess their accuracy. The number of SNPs called without a reference genome was lower (13k to 24k) than with a reference genome (25k to 54k SNPs) while accuracy was high (92.3 to 98.7%) for all but one pipeline (TASSEL-GBSv1, 76.1%). Among pipelines offering a high accuracy (>95%), Fast-GBS called the greatest number of polymorphisms (close to 35,000 SNPs + Indels) and yielded the highest accuracy (98.7%). Using Ion Torrent sequence data for the same 24 lines, we compared the performance of Fast-GBS with that of TASSEL-GBSv2. It again called more polymorphisms (25.8K vs 22.9K) and these proved more accurate (95.2 vs 91.1%). Typically, SNP catalogues called from the same sequencing data using different pipelines resulted in highly overlapping SNP catalogues (79-92% overlap). In contrast, overlap between SNP catalogues obtained using the same pipeline but different sequencing technologies was less extensive (~50-70%).
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
