

H3K36 methyltransferases as cancer drug targets: rationale and perspectives for inhibitor development
- PMID: 27548565
- PMCID: PMC5020427
- DOI: 10.4155/fmc-2016-0071
H3K36 methyltransferases as cancer drug targets: rationale and perspectives for inhibitor development
Abstract
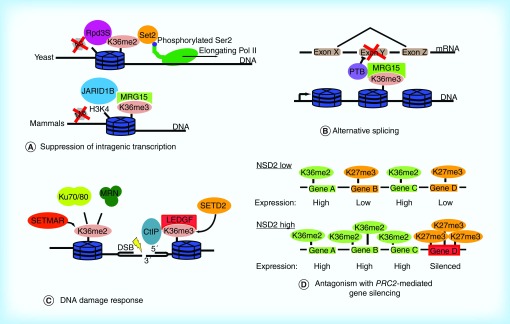
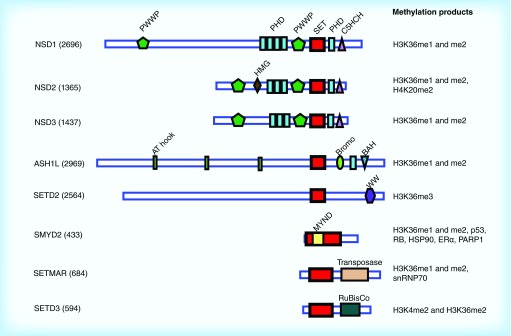
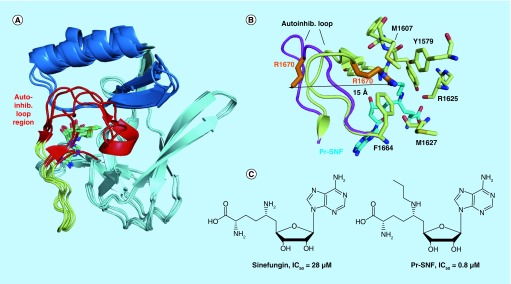
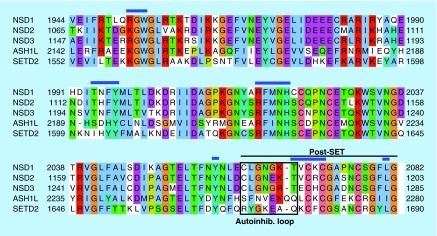
Methylation at histone 3, lysine 36 (H3K36) is a conserved epigenetic mark regulating gene transcription, alternative splicing and DNA repair. Genes encoding H3K36 methyltransferases (KMTases) are commonly overexpressed, mutated or involved in chromosomal translocations in cancer. Molecular biology studies have demonstrated that H3K36 KMTases regulate oncogenic transcriptional programs. Structural studies of the catalytic SET domain of H3K36 KMTases have revealed intriguing opportunities for design of small molecule inhibitors. Nevertheless, potent inhibitors for most H3K36 KMTases have not yet been developed, underlining the challenges associated with this target class. As we now have strong evidence linking H3K36 KMTases to cancer, drug development efforts are predicted to yield novel compounds in the near future.
Keywords: cancer; chromosomal translocation; epigenetics; gene expression; genome integrity; histone methyltransferase; oncogenes; protein–protein interactions; structure-based drug design.
Conflict of interest statement
Financial & competing interests disclosure This research has been supported by the Leukemia and Lymphoma Society TRP grants 6111-14 to T Cierpicki and 6485-16 to J Grembecka, NIH 1R01CA160467 to J Grembecka and 1R01CA181185 to T Cierpicki. J Grembecka and T Cierpicki are Leukemia and Lymphoma Society Scholars (grants 1215-14 and 1340-17). DS Rogawski acknowledges training grant support from the University of Michigan Chemistry–Biology Interface (CBI) training program (NIH Grant 5T32GM008597) and from the University of Michigan Medical Scientist Training Program (NIH Grant 5T32GM007863). J Grembecka and T Cierpicki receive research support from Kura Oncology. They are also receiving compensation as members of the scientific advisory board of Kura Oncology, and they have an equity ownership in the company. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.
Figures
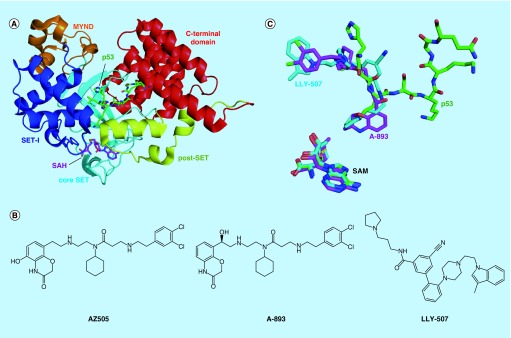
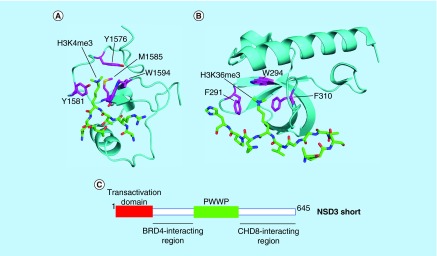
References
-
- Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150(1):12–27. - PubMed
-
- McCabe MT, Creasy CL. EZH2 as a potential target in cancer therapy. Epigenomics. 2014;6(3):341–351. - PubMed
-
- Waters NJ, Smith SA, Olhava EJ, et al. Metabolism and disposition of the DOT1L inhibitor, pinometostat (EPZ-5676), in rat, dog and human. Cancer Chemother. Pharmacol. 2016;77(1):43–62. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials