

Reduction of MDSCs with All-trans Retinoic Acid Improves CAR Therapy Efficacy for Sarcomas
- PMID: 27549124
- PMCID: PMC5050151
- DOI: 10.1158/2326-6066.CIR-15-0230
Reduction of MDSCs with All-trans Retinoic Acid Improves CAR Therapy Efficacy for Sarcomas
Abstract
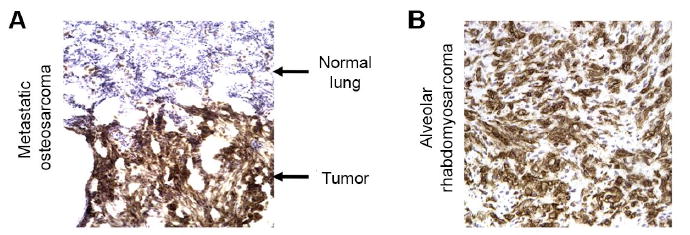
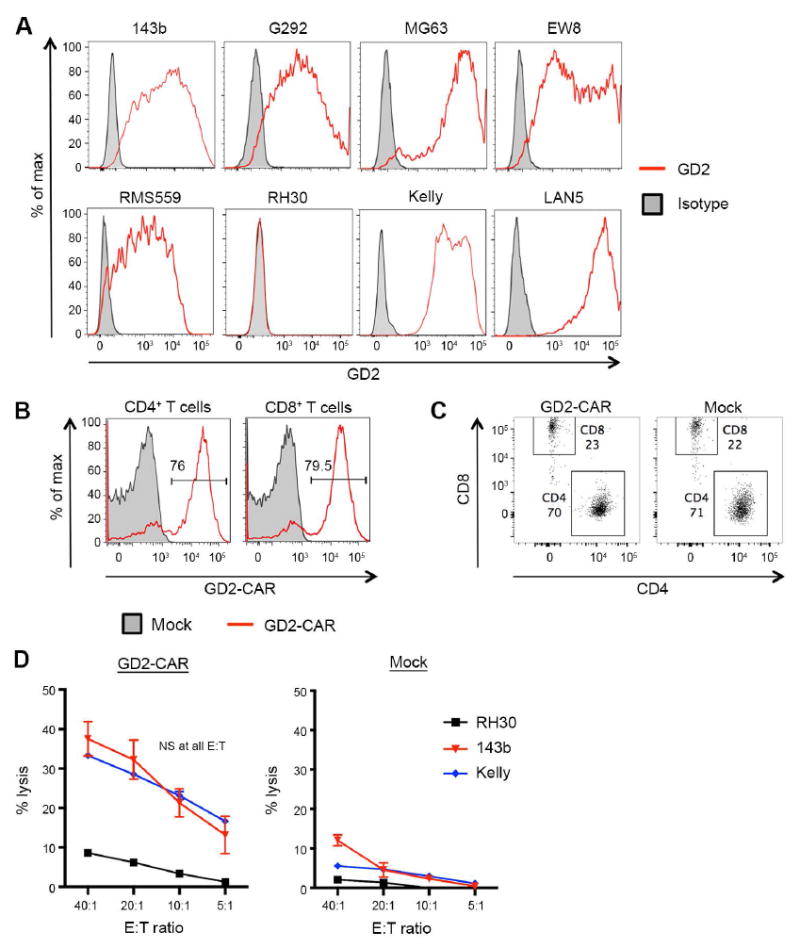
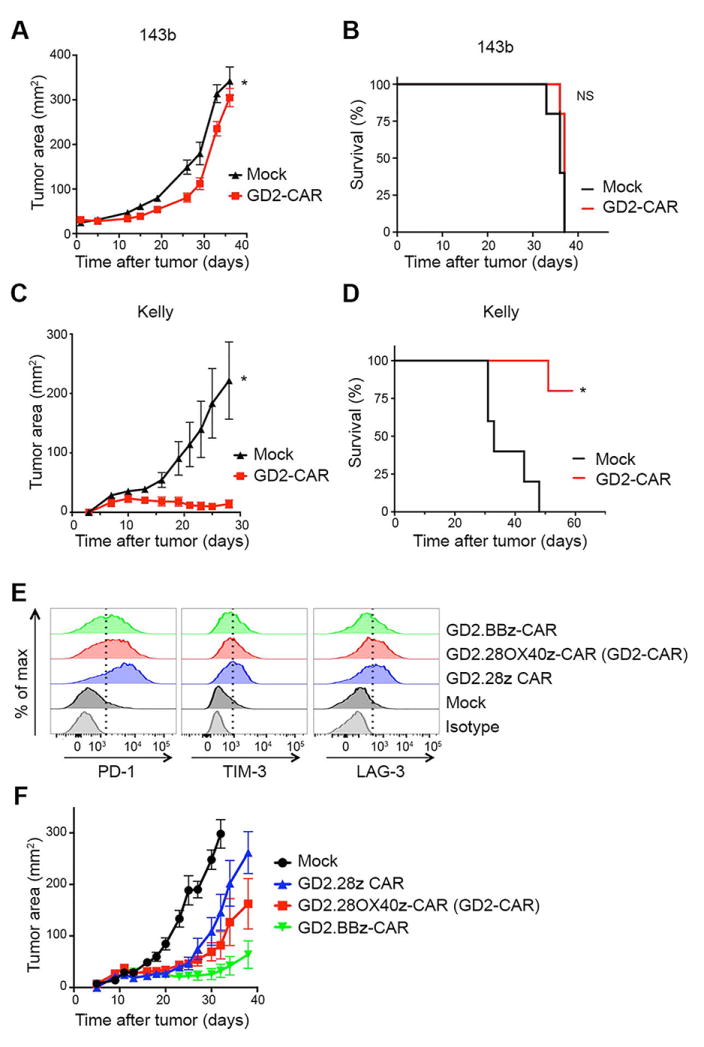
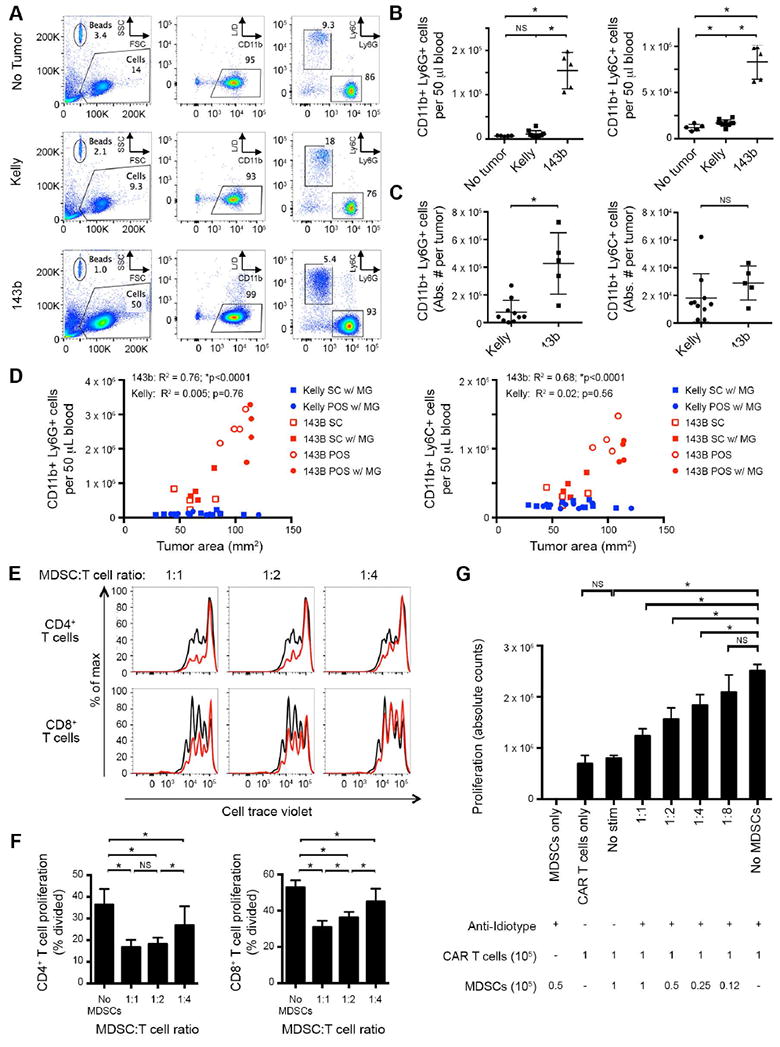
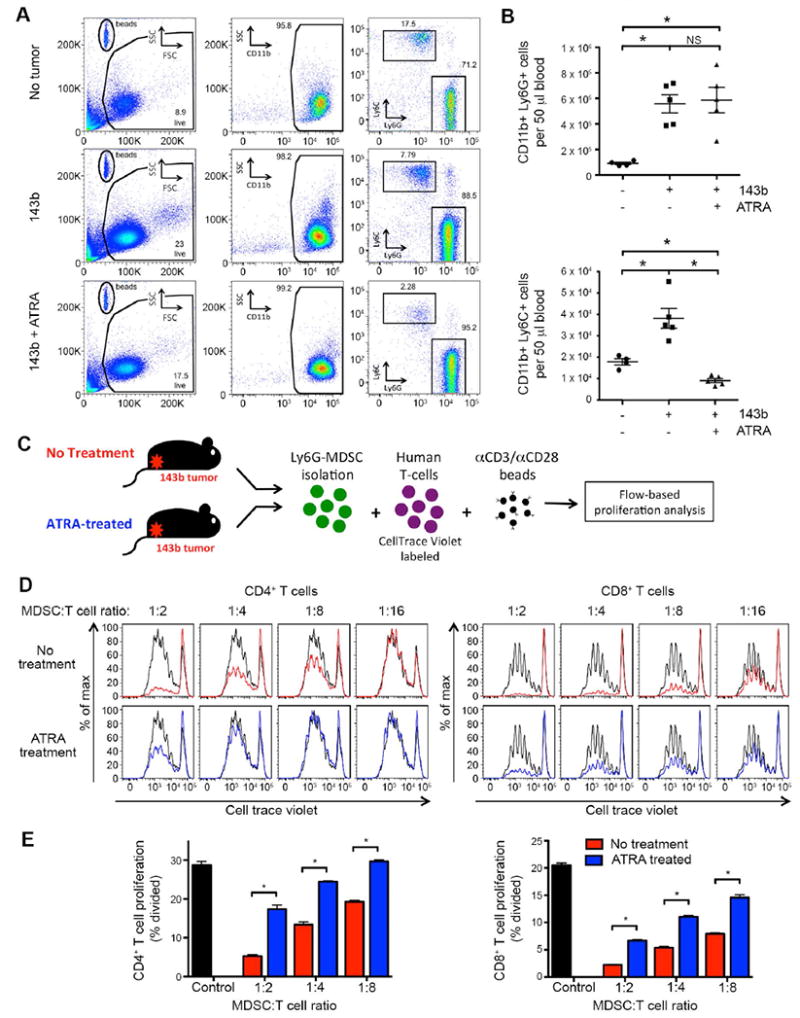
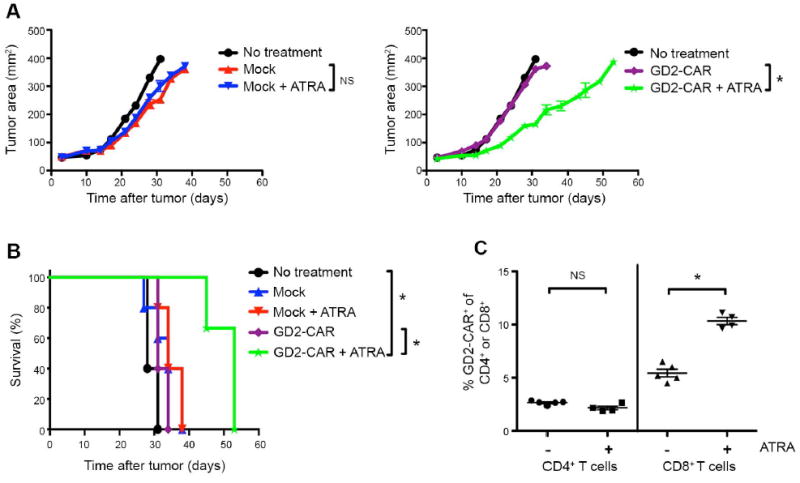
Genetically engineered T cells expressing CD19-specific chimeric antigen receptors (CAR) have shown impressive activity against B-cell malignancies, and preliminary results suggest that T cells expressing a first-generation disialoganglioside (GD2)-specific CAR can also provide clinical benefit in patients with neuroblastoma. We sought to assess the potential of GD2-CAR therapies to treat pediatric sarcomas. We observed that 18 of 18 (100%) of osteosarcomas, 2 of 15 (13%) of rhabdomyosarcomas, and 7 of 35 (20%) of Ewing sarcomas expressed GD2. T cells engineered to express a third-generation GD2-CAR incorporating the 14g2a-scFv with the CD28, OX40, and CD3ζ signaling domains (14g2a.CD28.OX40.ζ) mediated efficient and comparable lysis of both GD2+ sarcoma and neuroblastoma cell lines in vitro However, in xenograft models, GD2-CAR T cells had no antitumor effect against GD2+ sarcoma, despite effectively controlling GD2+ neuroblastoma. We observed that pediatric sarcoma xenografts, but not neuroblastoma xenografts, induced large populations of monocytic and granulocytic murine myeloid-derived suppressor cells (MDSC) that inhibited human CAR T-cell responses in vitro Treatment of sarcoma-bearing mice with all-trans retinoic acid (ATRA) largely eradicated monocytic MDSCs and diminished the suppressive capacity of granulocytic MDSCs. Combined therapy using GD2-CAR T cells plus ATRA significantly improved antitumor efficacy against sarcoma xenografts. We conclude that retinoids provide a clinically accessible class of agents capable of diminishing the suppressive effects of MDSCs, and that co-administration of retinoids may enhance the efficacy of CAR therapies targeting solid tumors. Cancer Immunol Res; 4(10); 869-80. ©2016 AACR.
©2016 American Association for Cancer Research.
Conflict of interest statement
The authors report no conflict of interest exists.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
