

Mapping transiently formed and sparsely populated conformations on a complex energy landscape
- PMID: 27552057
- PMCID: PMC5050026
- DOI: 10.7554/eLife.17505
Mapping transiently formed and sparsely populated conformations on a complex energy landscape
Abstract
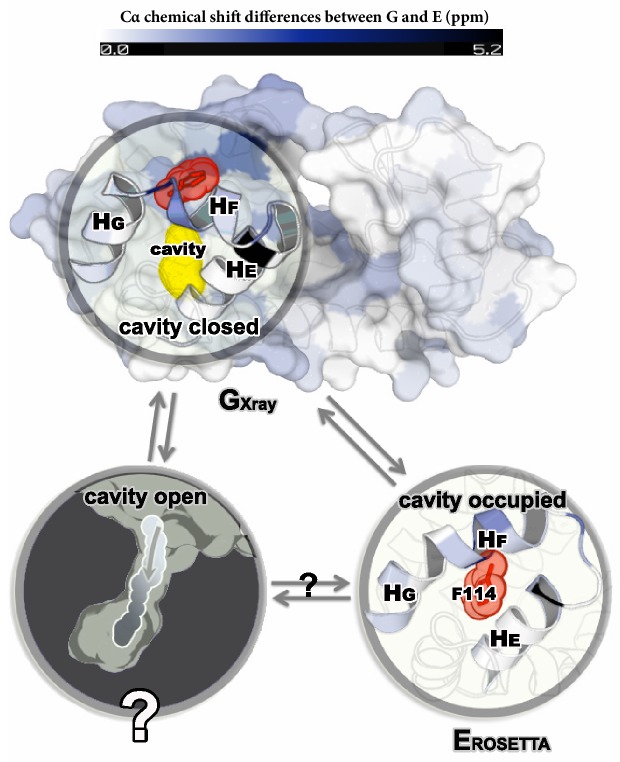
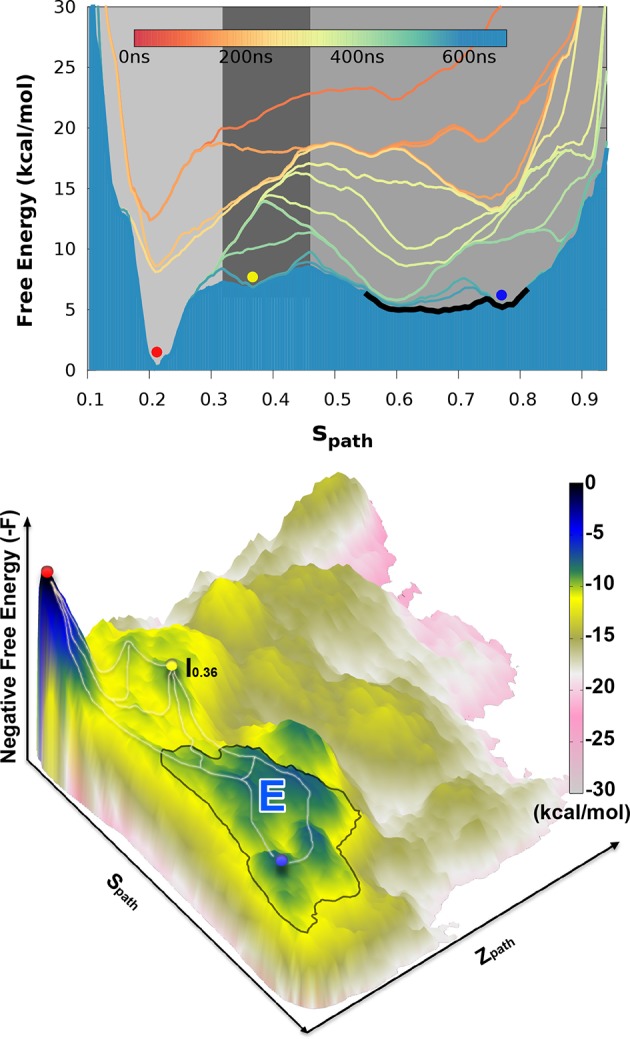
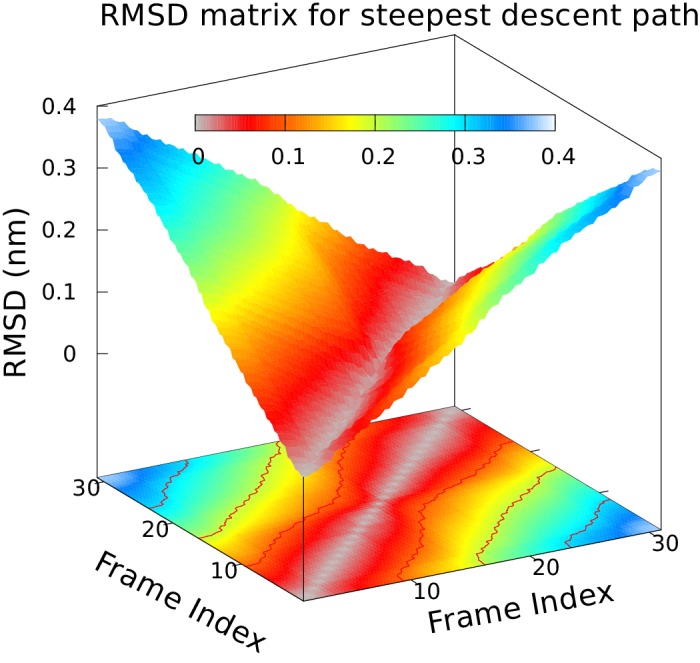
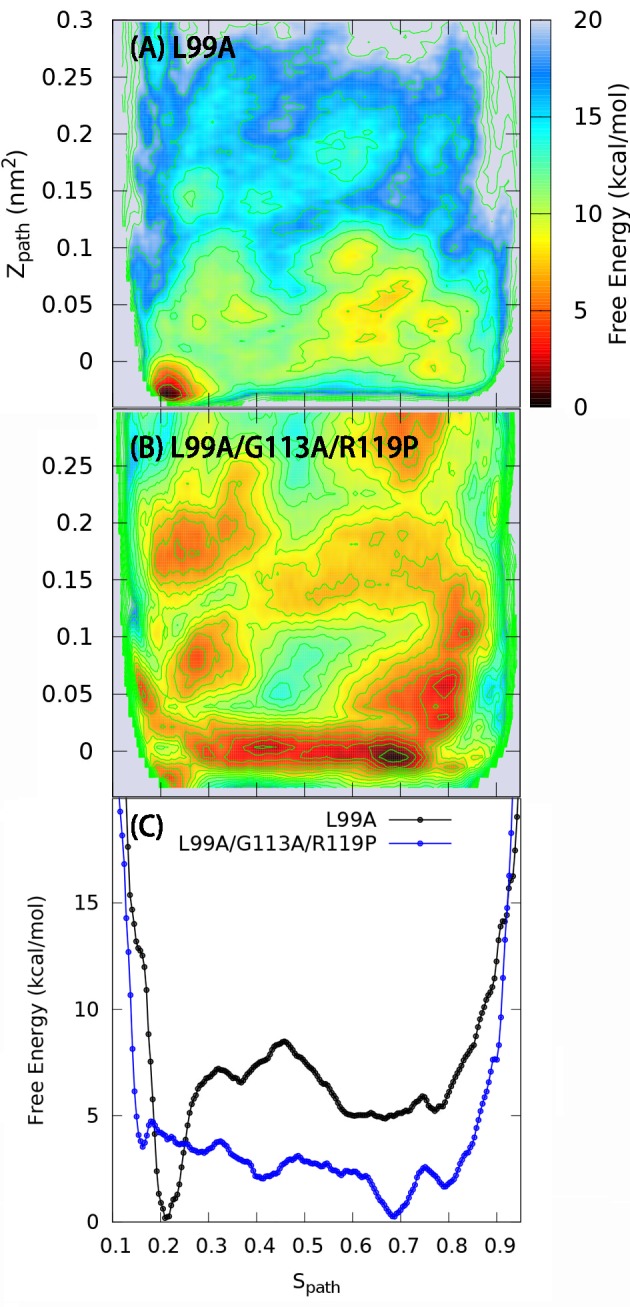
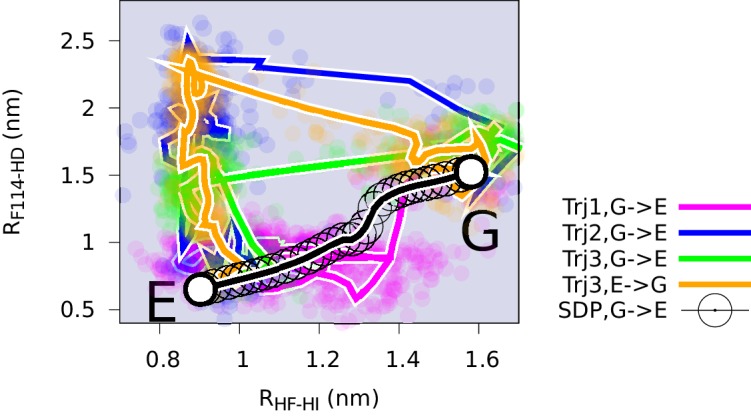
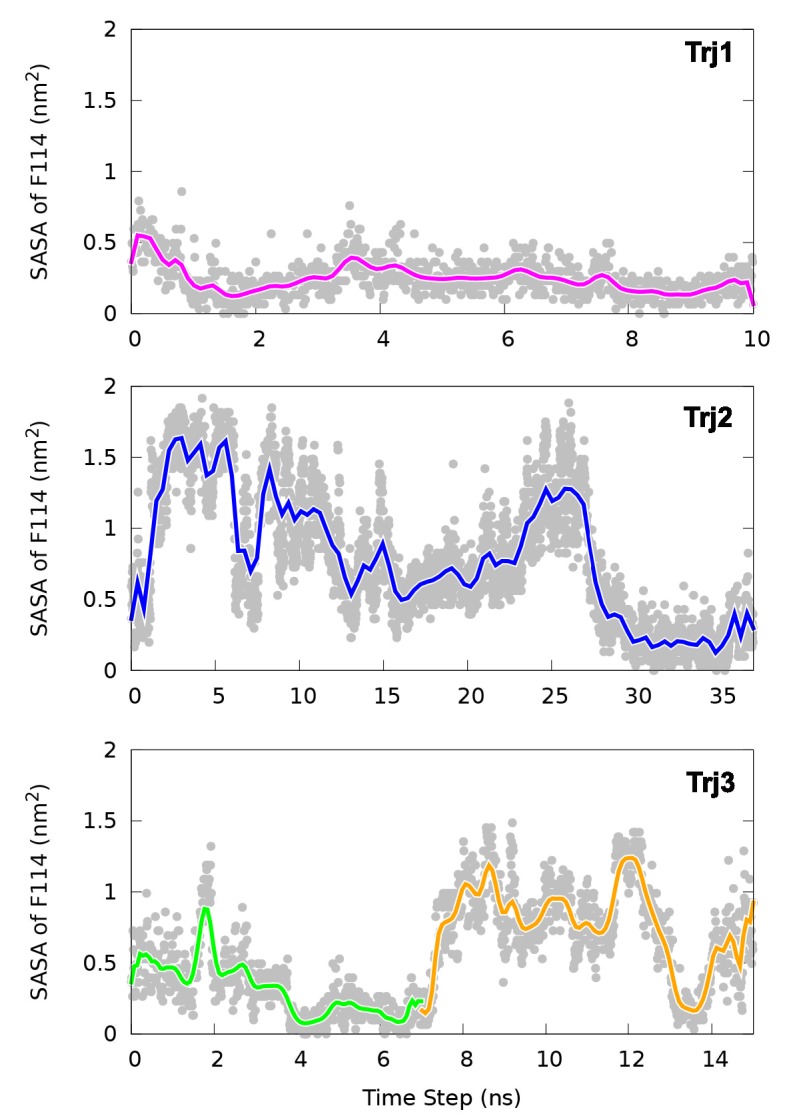
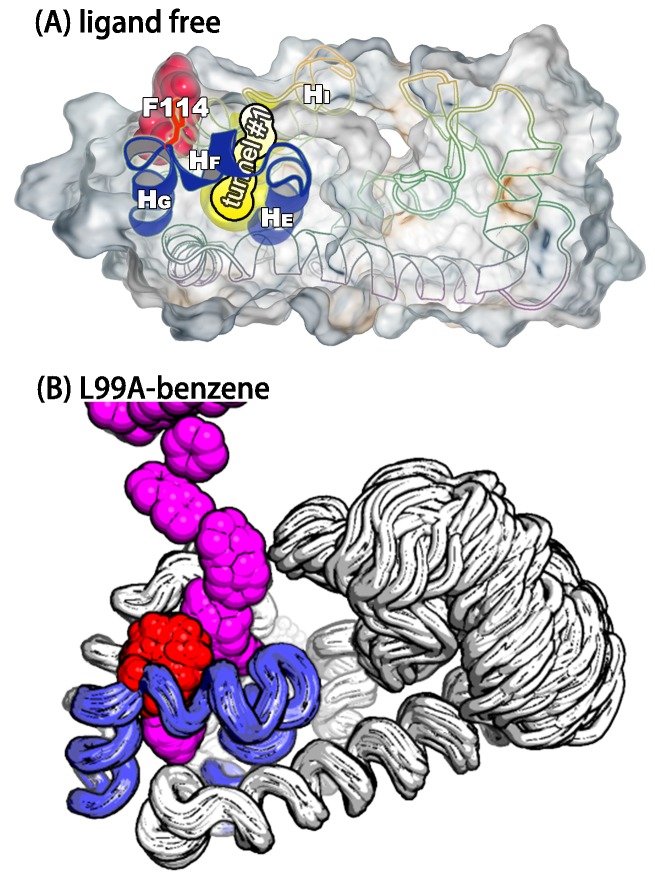
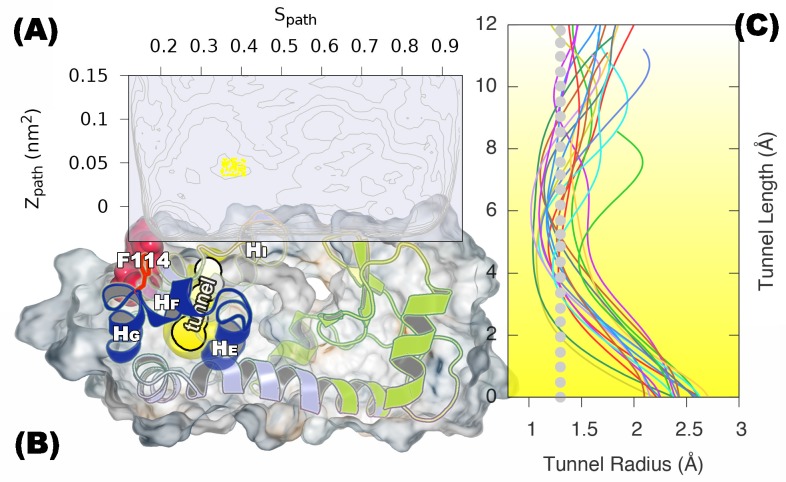
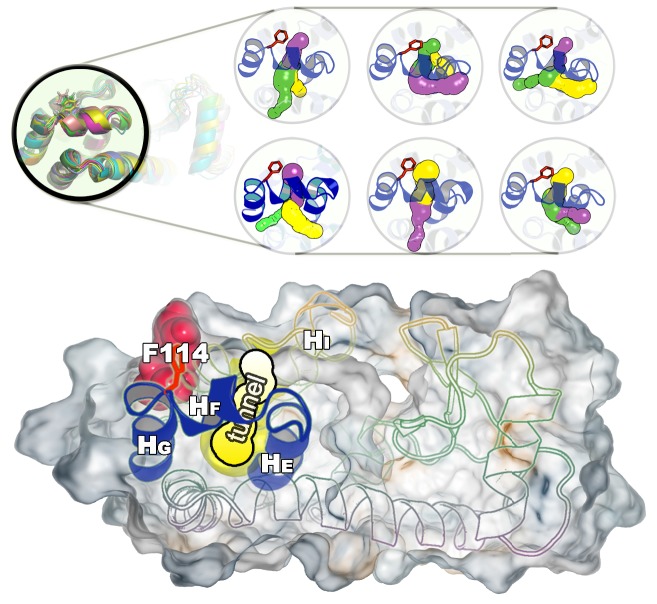
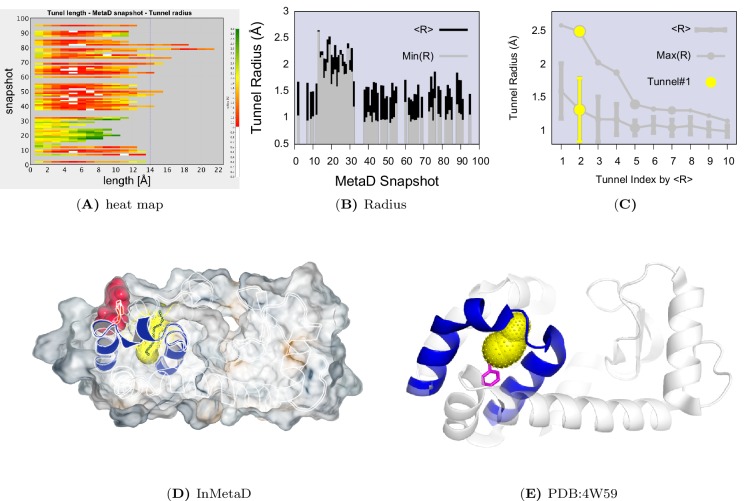
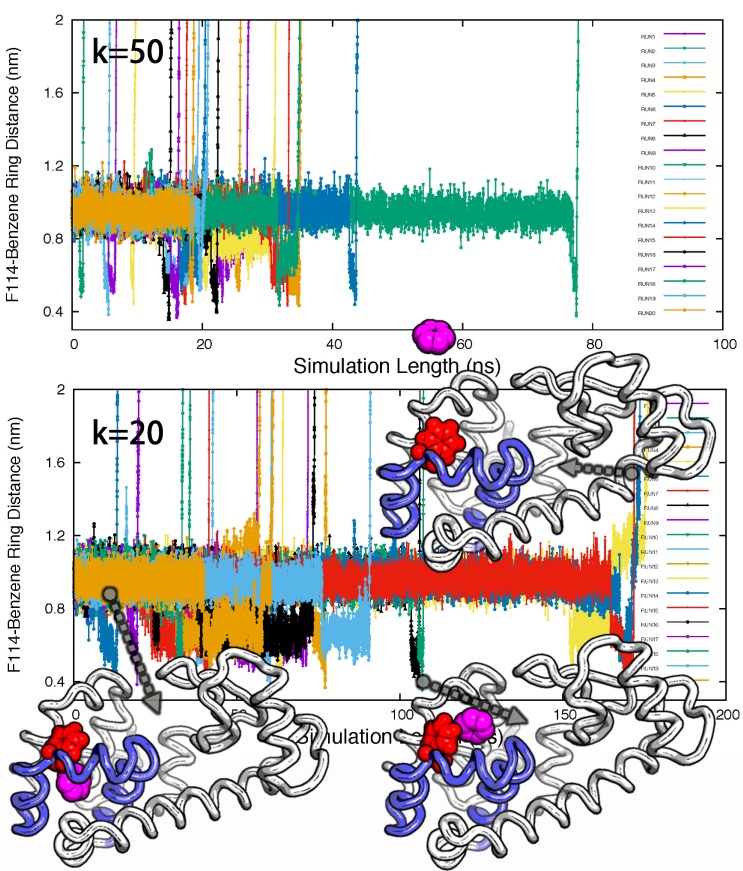
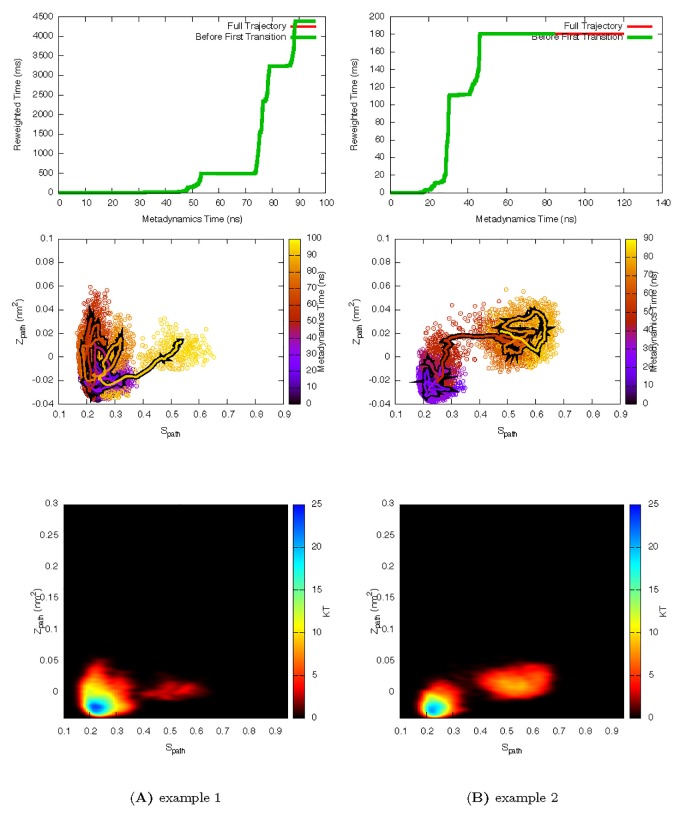
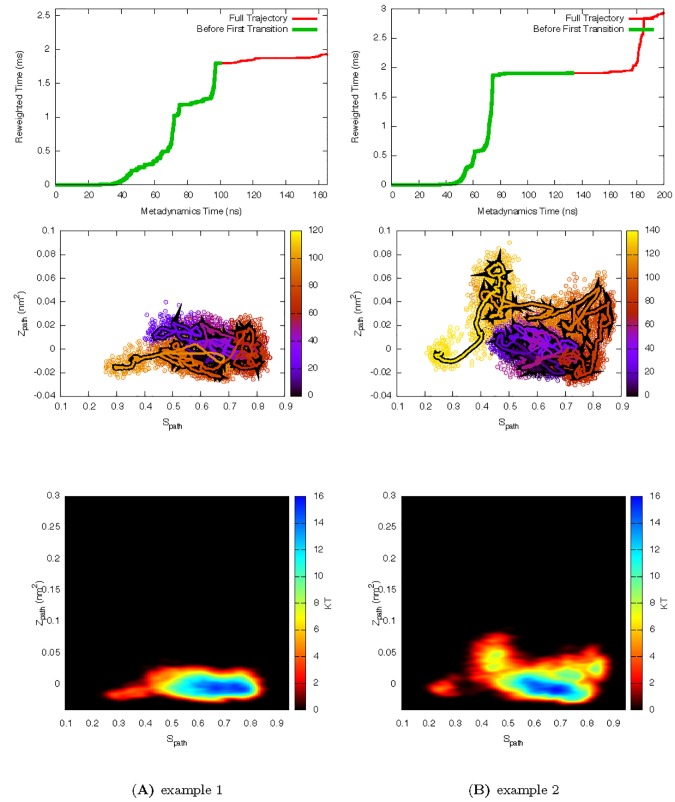
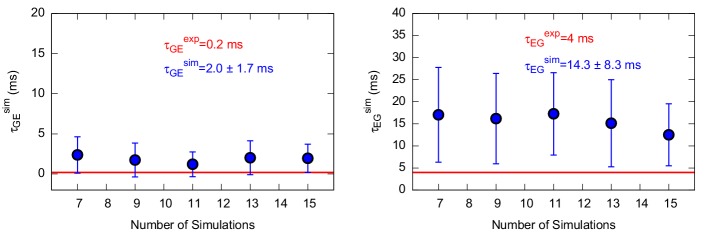
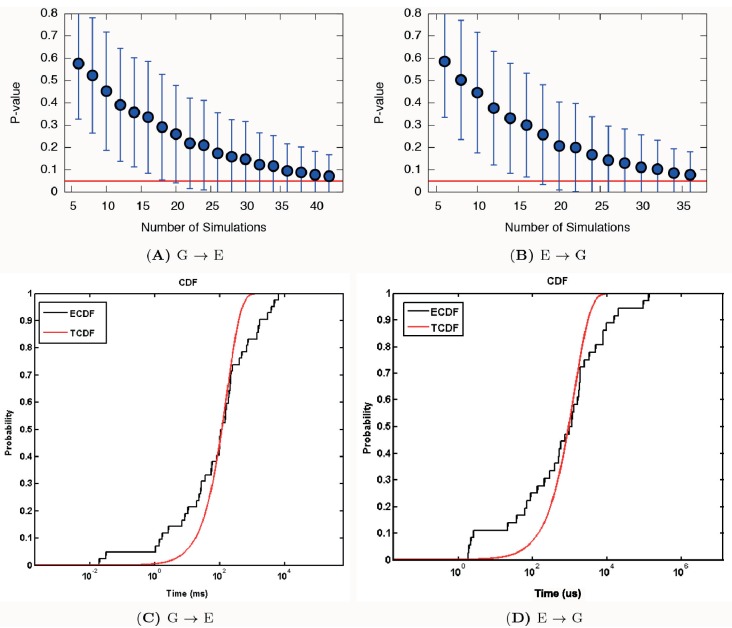
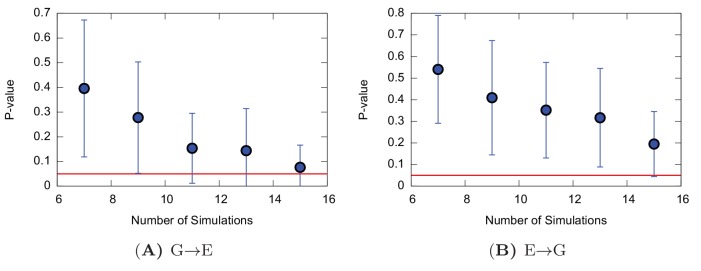
Determining the structures, kinetics, thermodynamics and mechanisms that underlie conformational exchange processes in proteins remains extremely difficult. Only in favourable cases is it possible to provide atomic-level descriptions of sparsely populated and transiently formed alternative conformations. Here we benchmark the ability of enhanced-sampling molecular dynamics simulations to determine the free energy landscape of the L99A cavity mutant of T4 lysozyme. We find that the simulations capture key properties previously measured by NMR relaxation dispersion methods including the structure of a minor conformation, the kinetics and thermodynamics of conformational exchange, and the effect of mutations. We discover a new tunnel that involves the transient exposure towards the solvent of an internal cavity, and show it to be relevant for ligand escape. Together, our results provide a comprehensive view of the structural landscape of a protein, and point forward to studies of conformational exchange in systems that are less characterized experimentally.
Keywords: biochemistry; biophysics; conformational exchange; free energy landscape; kinetics; molecular dynamics; nuclear magnetic resonance; structural biology.
Conflict of interest statement
The authors declare that no competing interests exist.
Figures

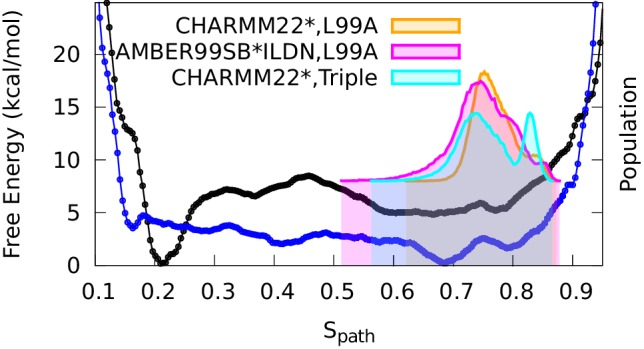
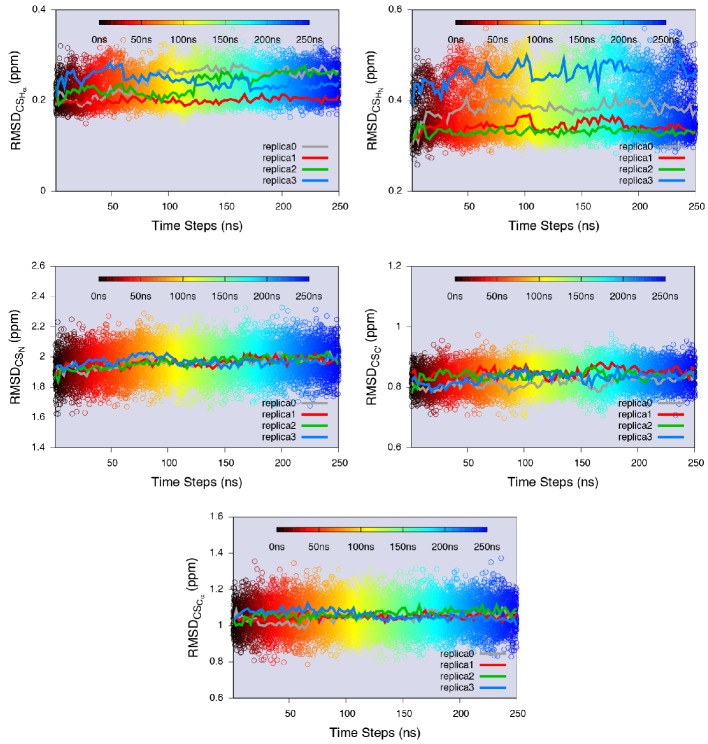
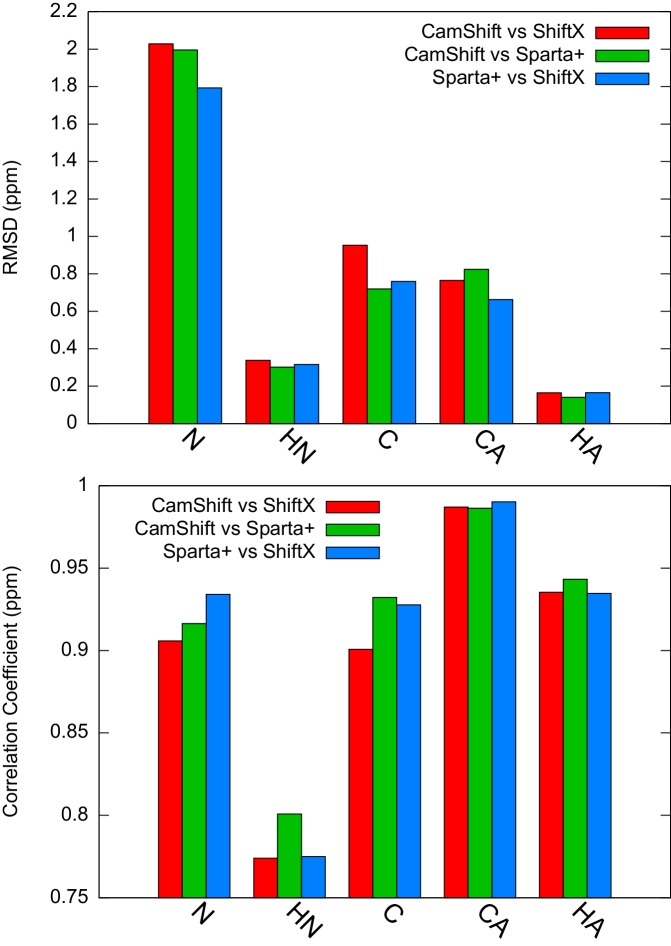
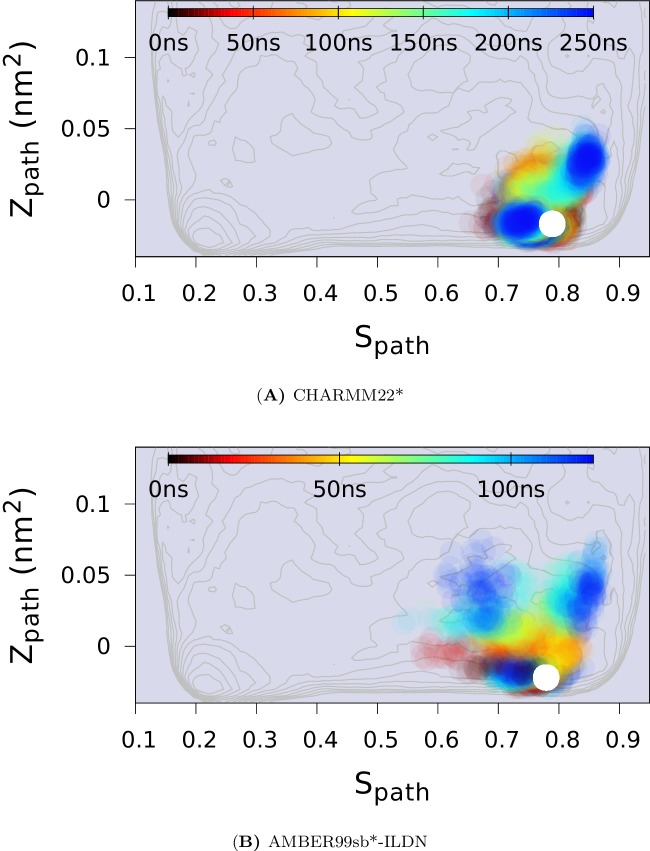
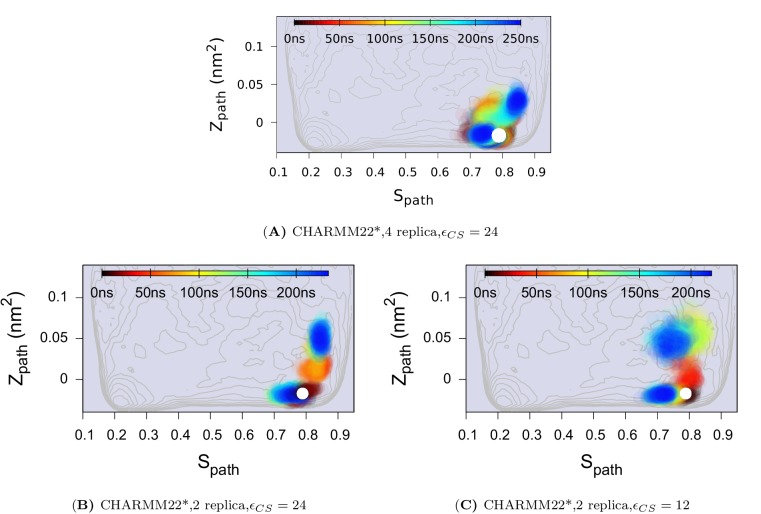
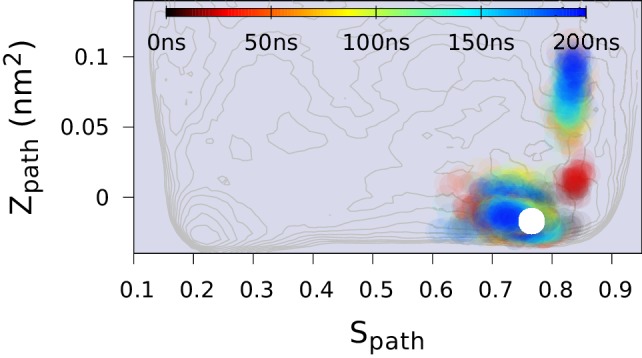
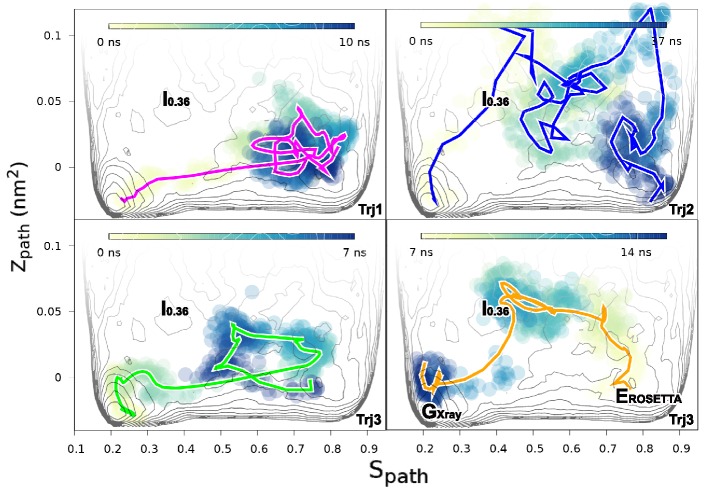
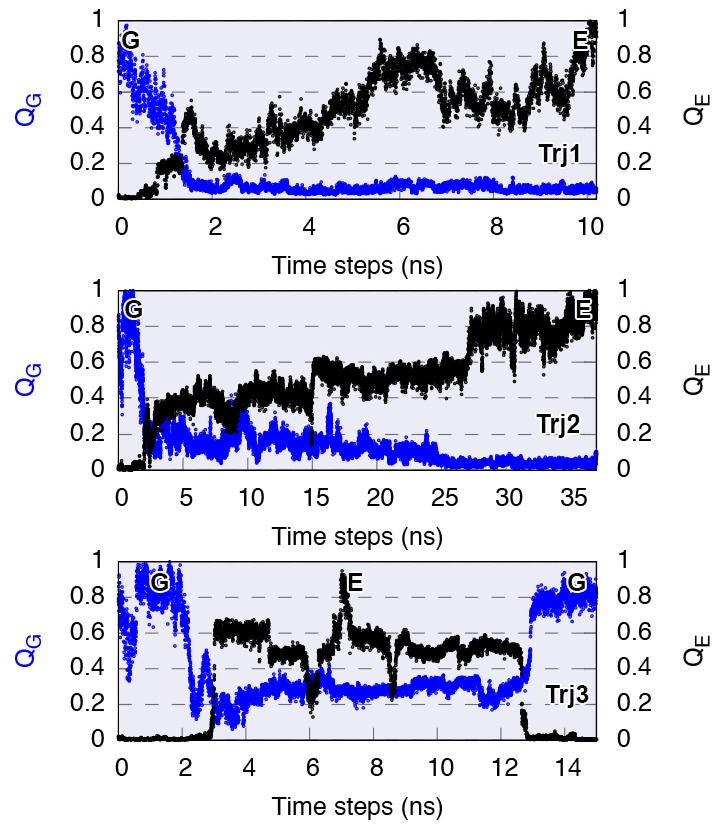

References
-
- Barducci A, Pfaendtner J, Bonomi M. Molecular Modeling of Proteins. Springer; 2015. Tackling sampling challenges in biomolecular simulations; pp. 151–171. - PubMed
-
- Bonomi M, Branduardi D, Bussi G, Camilloni C, Provasi D, Raiteri P, Donadio D, Marinelli F, Pietrucci F, Broglia RA, Parrinello M. PLUMED: A portable plugin for free-energy calculations with molecular dynamics. Computer Physics Communications. 2009;180:1961–1972. doi: 10.1016/j.cpc.2009.05.011. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
