

MuSE: accounting for tumor heterogeneity using a sample-specific error model improves sensitivity and specificity in mutation calling from sequencing data
- PMID: 27557938
- PMCID: PMC4995747
- DOI: 10.1186/s13059-016-1029-6
MuSE: accounting for tumor heterogeneity using a sample-specific error model improves sensitivity and specificity in mutation calling from sequencing data
Abstract
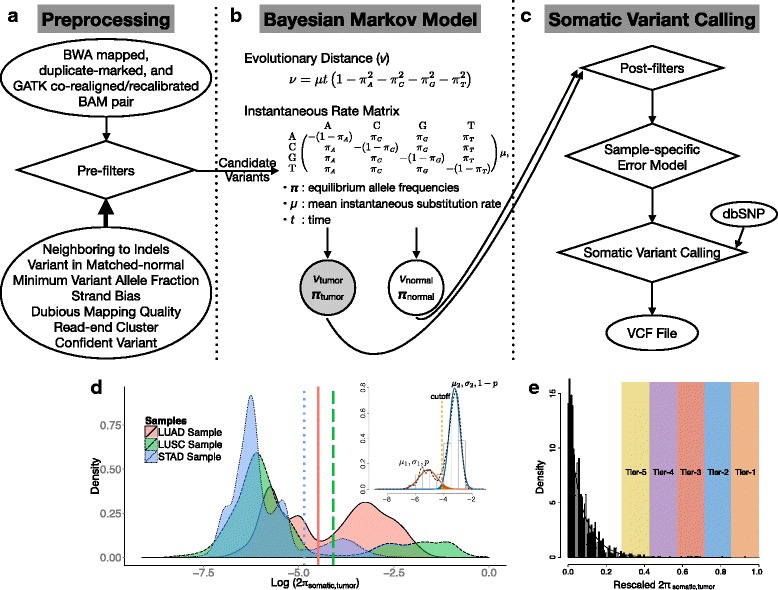
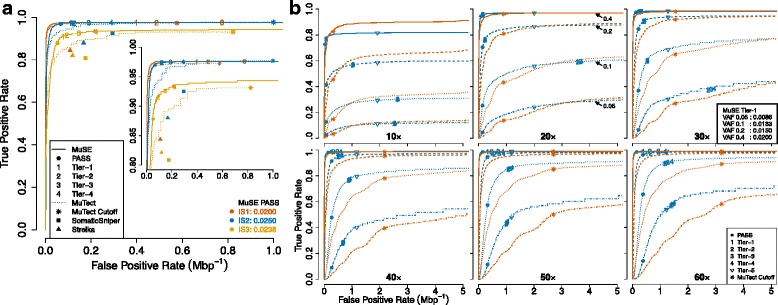
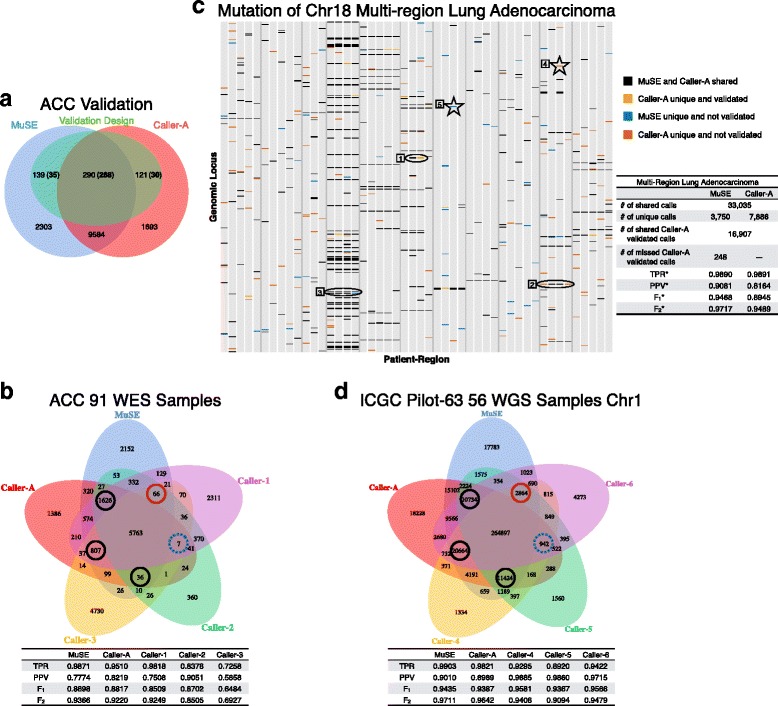
Subclonal mutations reveal important features of the genetic architecture of tumors. However, accurate detection of mutations in genetically heterogeneous tumor cell populations using next-generation sequencing remains challenging. We develop MuSE ( http://bioinformatics.mdanderson.org/main/MuSE ), Mutation calling using a Markov Substitution model for Evolution, a novel approach for modeling the evolution of the allelic composition of the tumor and normal tissue at each reference base. MuSE adopts a sample-specific error model that reflects the underlying tumor heterogeneity to greatly improve the overall accuracy. We demonstrate the accuracy of MuSE in calling subclonal mutations in the context of large-scale tumor sequencing projects using whole exome and whole genome sequencing.
Keywords: Bayesian inference; Model-based cutoff finding; Next-generation sequencing; Sensitivity and specificity; Somatic mutation calling.
Figures
References
-
- Roth A, Ding J, Morin R, Crisan A, Ha G, Giuliany R, et al. JointSNVMix: a probabilistic model for accurate detection of somatic mutations in normal/tumour paired next-generation sequencing data. Bioinformatics (Oxford, England) 2012;28(7):907–13. doi: 10.1093/bioinformatics/bts053. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
