

A generic, cost-effective, and scalable cell lineage analysis platform
- PMID: 27558250
- PMCID: PMC5088600
- DOI: 10.1101/gr.202903.115
A generic, cost-effective, and scalable cell lineage analysis platform
Abstract
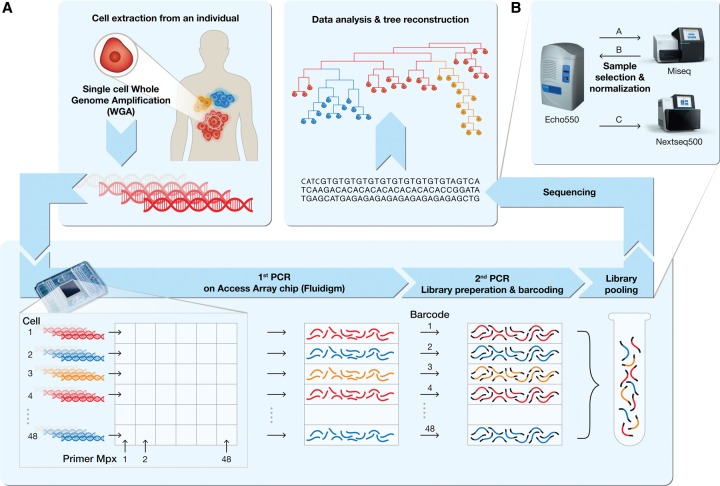
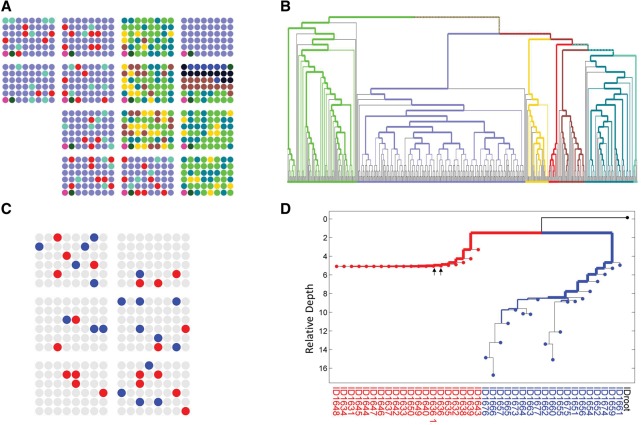
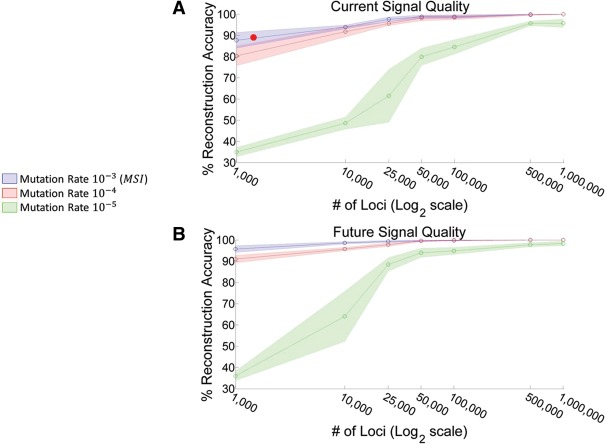
Advances in single-cell genomics enable commensurate improvements in methods for uncovering lineage relations among individual cells. Current sequencing-based methods for cell lineage analysis depend on low-resolution bulk analysis or rely on extensive single-cell sequencing, which is not scalable and could be biased by functional dependencies. Here we show an integrated biochemical-computational platform for generic single-cell lineage analysis that is retrospective, cost-effective, and scalable. It consists of a biochemical-computational pipeline that inputs individual cells, produces targeted single-cell sequencing data, and uses it to generate a lineage tree of the input cells. We validated the platform by applying it to cells sampled from an ex vivo grown tree and analyzed its feasibility landscape by computer simulations. We conclude that the platform may serve as a generic tool for lineage analysis and thus pave the way toward large-scale human cell lineage discovery.
© 2016 Biezuner et al.; Published by Cold Spring Harbor Laboratory Press.
Figures


References
-
- Boyer JC, Umar A, Risinger JI, Lipford JR, Kane M, Yin S, Barrett JC, Kolodner RD, Kunkel TA. 1995. Microsatellite instability, mismatch repair deficiency, and genetic defects in human cancer cell lines. Cancer Res 55: 6063–6070. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources