

Whole-genome characterization in pedigreed non-human primates using genotyping-by-sequencing (GBS) and imputation
- PMID: 27558348
- PMCID: PMC4997765
- DOI: 10.1186/s12864-016-2966-x
Whole-genome characterization in pedigreed non-human primates using genotyping-by-sequencing (GBS) and imputation
Abstract
Background: Rhesus macaques are widely used in biomedical research, but the application of genomic information in this species to better understand human disease is still in its infancy. Whole-genome sequence (WGS) data in large pedigreed macaque colonies could provide substantial experimental power for genetic discovery, but the collection of WGS data in large cohorts remains a formidable expense. Here, we describe a cost-effective approach that selects the most informative macaques in a pedigree for 30X WGS, followed by low-cost genotyping-by-sequencing (GBS) at 30X on the remaining macaques in order to generate sparse genotype data at high accuracy. Dense variants from the selected macaques with WGS data are then imputed into macaques having only sparse GBS data, resulting in dense genome-wide genotypes throughout the pedigree.
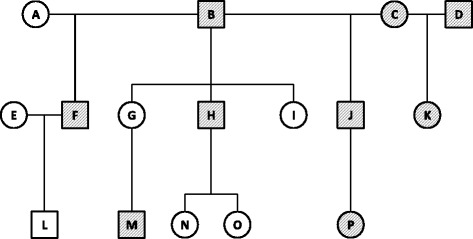
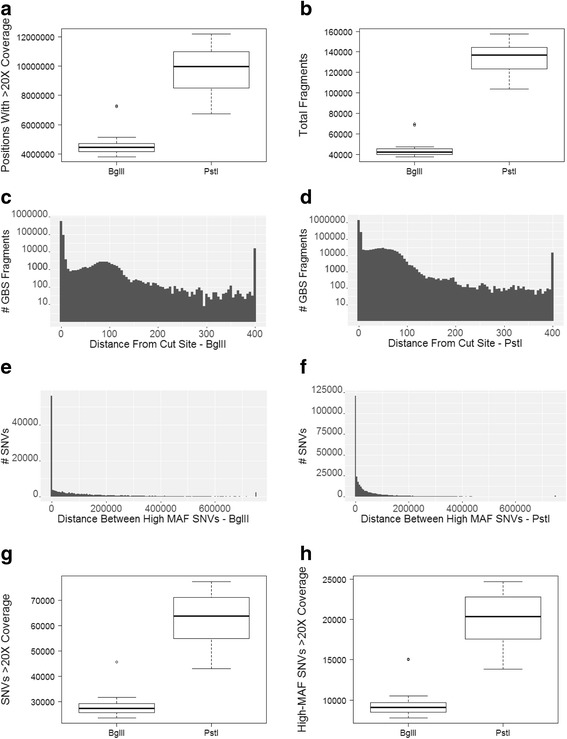
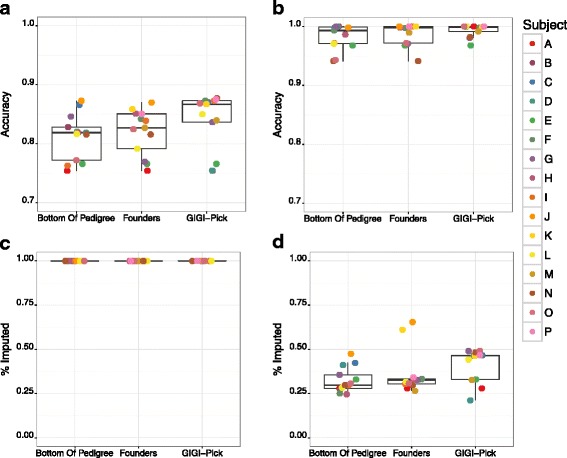
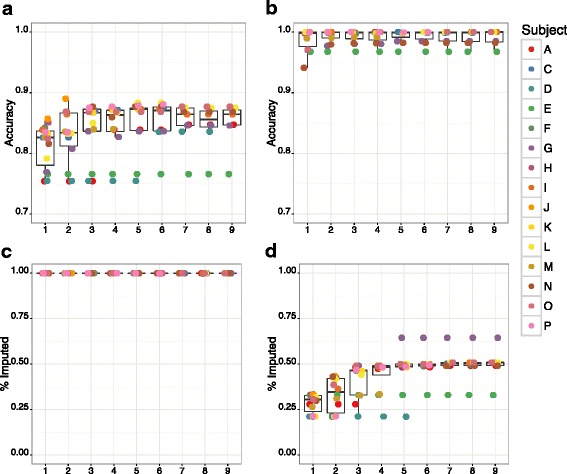
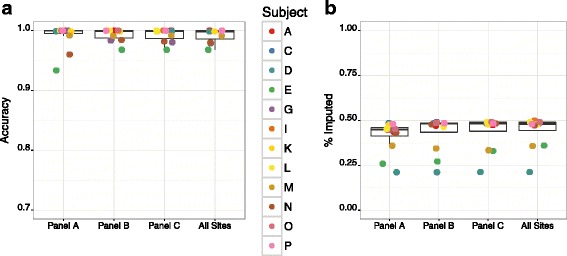
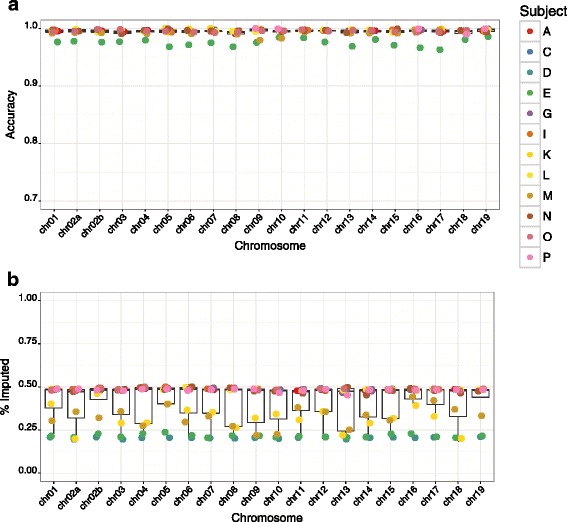
Results: We developed GBS for the macaque genome using a digestion with PstI, followed by sequencing of size-selected fragments at 30X coverage. From GBS sequence data collected on all individuals in a 16-member pedigree, we characterized high-confidence genotypes at 22,455 single nucleotide variant (SNV) sites that were suitable for guiding imputation of dense sequence data from WGS. To characterize dense markers for imputation, we performed WGS at 30X coverage on nine of the 16 individuals, yielding 10,193,425 high-confidence SNVs. To validate the use of GBS data for facilitating imputation, we initially focused on chromosome 19 as a test case, using an optimized panel of 833 sparse, evenly-spaced markers from GBS and 5,010 dense markers from WGS. Using the method of "Genotype Imputation Given Inheritance" (GIGI), we evaluated the effects on imputation accuracy of 3 different strategies for selecting individuals for WGS, including 1) using "GIGI-Pick" to select the most informative individuals, 2) using the most recent generation, or 3) using founders only. We also evaluated the effects on imputation accuracy of using a range of from 1 to 9 WGS individuals for imputation. We found that the GIGI-Pick algorithm for selection of WGS individuals outperformed common heuristic approaches, and that genotype numbers and accuracy improved very little when using >5 WGS individuals for imputation. Informed by our findings, we used 4 macaques with WGS data to impute variants at up to 7,655,491 sites spanning all 20 autosomes in the 12 remaining macaques, based on their GBS genotypes at only 17,158 loci. Using a strict confidence threshold, we imputed an average of 3,680,238 variants per individual at >99 % accuracy, or an average 4,458,883 variants per individual at a more relaxed threshold, yielding >97 % accuracy.
Conclusions: We conclude that an optimal tradeoff between genotype accuracy, number of imputed genotypes, and overall cost exists at the ratio of one individual selected for WGS using the GIGI-Pick algorithm, per 3-5 relatives selected for GBS. This approach makes feasible the collection of accurate, dense genome-wide sequence data in large pedigreed macaque cohorts without the need for more expensive WGS data on all individuals.
Keywords: Whole-genome sequencing; genotyping-by-sequencing; imputation; macaque; pedigree.
Figures
References
-
- Bielenberg DG, Rauh B, Fan S, Gasic K, Abbott AG, Reighard GL, Okie WR, Wells CE. Genotyping by Sequencing for SNP-Based Linkage Map Construction and QTL Analysis of Chilling Requirement and Bloom Date in Peach [Prunus persica (L.) Batsch] PLoS One. 2015;10(10):e0139406. doi: 10.1371/journal.pone.0139406. - DOI - PMC - PubMed
-
- Palti Y, Vallejo RL, Gao G, Liu S, Hernandez AG, Rexroad CE, 3rd, Wiens GD. Detection and Validation of QTL Affecting Bacterial Cold Water Disease Resistance in Rainbow Trout Using Restriction-Site Associated DNA Sequencing. PLoS One. 2015;10(9):e0138435. doi: 10.1371/journal.pone.0138435. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
