

Navigating Microbiological Food Safety in the Era of Whole-Genome Sequencing
- PMID: 27559074
- PMCID: PMC5010751
- DOI: 10.1128/CMR.00056-16
Navigating Microbiological Food Safety in the Era of Whole-Genome Sequencing
Abstract
The epidemiological investigation of a foodborne outbreak, including identification of related cases, source attribution, and development of intervention strategies, relies heavily on the ability to subtype the etiological agent at a high enough resolution to differentiate related from nonrelated cases. Historically, several different molecular subtyping methods have been used for this purpose; however, emerging techniques, such as single nucleotide polymorphism (SNP)-based techniques, that use whole-genome sequencing (WGS) offer a resolution that was previously not possible. With WGS, unlike traditional subtyping methods that lack complete information, data can be used to elucidate phylogenetic relationships and disease-causing lineages can be tracked and monitored over time. The subtyping resolution and evolutionary context provided by WGS data allow investigators to connect related illnesses that would be missed by traditional techniques. The added advantage of data generated by WGS is that these data can also be used for secondary analyses, such as virulence gene detection, antibiotic resistance gene profiling, synteny comparisons, mobile genetic element identification, and geographic attribution. In addition, several software packages are now available to generate in silico results for traditional molecular subtyping methods from the whole-genome sequence, allowing for efficient comparison with historical databases. Metagenomic approaches using next-generation sequencing have also been successful in the detection of nonculturable foodborne pathogens. This review addresses state-of-the-art techniques in microbial WGS and analysis and then discusses how this technology can be used to help support food safety investigations. Retrospective outbreak investigations using WGS are presented to provide organism-specific examples of the benefits, and challenges, associated with WGS in comparison to traditional molecular subtyping techniques.
© Crown copyright 2016.
Figures
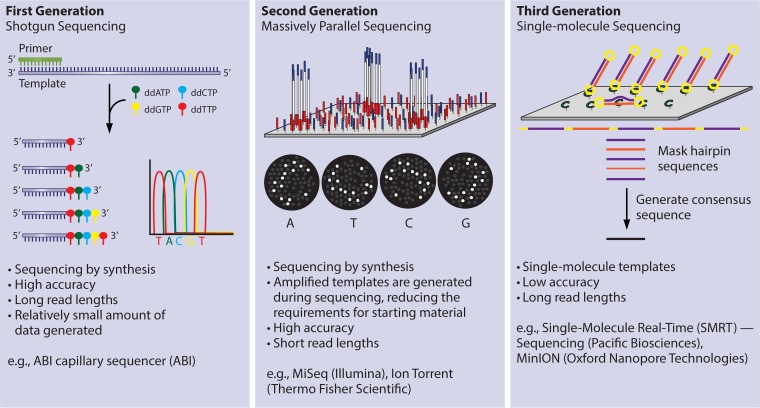
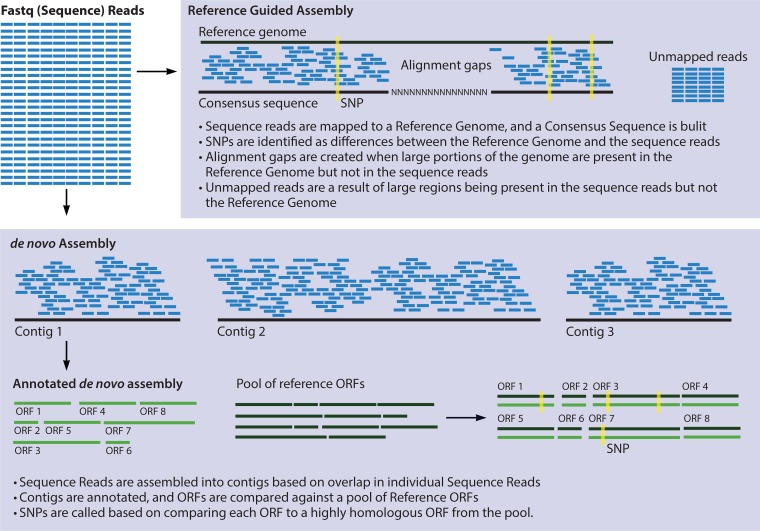
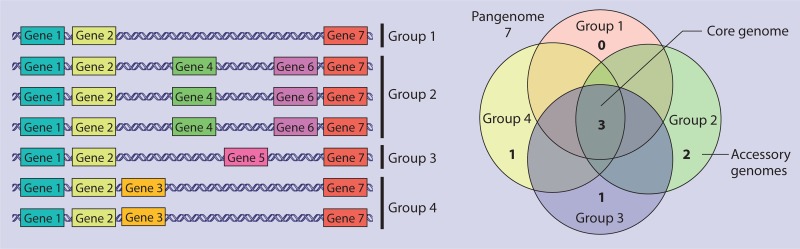
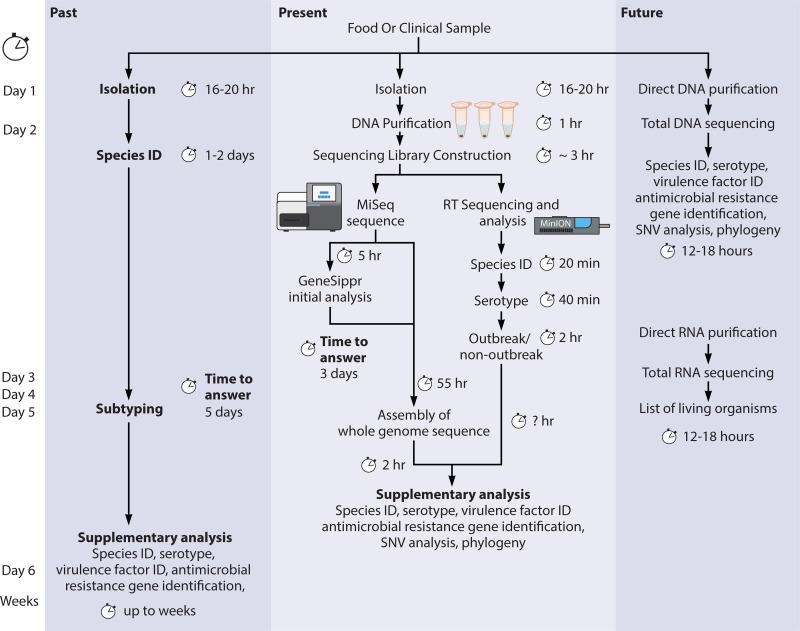




References
-
- Franz E, Gras LM, Dallman T. 2016. Significance of whole genome sequencing for surveillance, source attribution and microbial risk assessment of foodborne pathogens. Curr Opin Food Sci 8:74–79. doi: 10.1016/j.cofs.2016.04.004. - DOI
-
- Grimont PAD, Weill F-X. 2007. Antigenic formulae of the Salmonella serovars, 9th ed WHO Collaborating Centre for Reference and Research on Salmonella, Paris, France: http://www.scacm.org/free/Antigenic%20Formulae%20of%20the%20Salmonella%2....
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
