

Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease
- PMID: 27559085
- PMCID: PMC5018768
- DOI: 10.1073/pnas.1612594113
Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease
Abstract
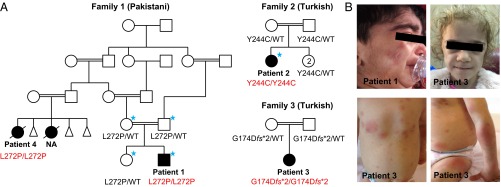
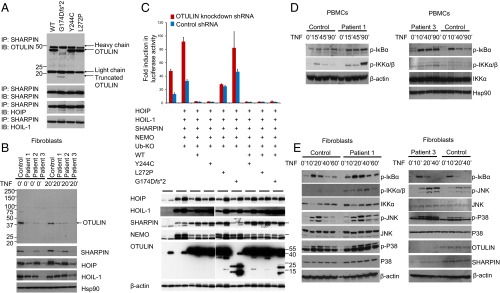
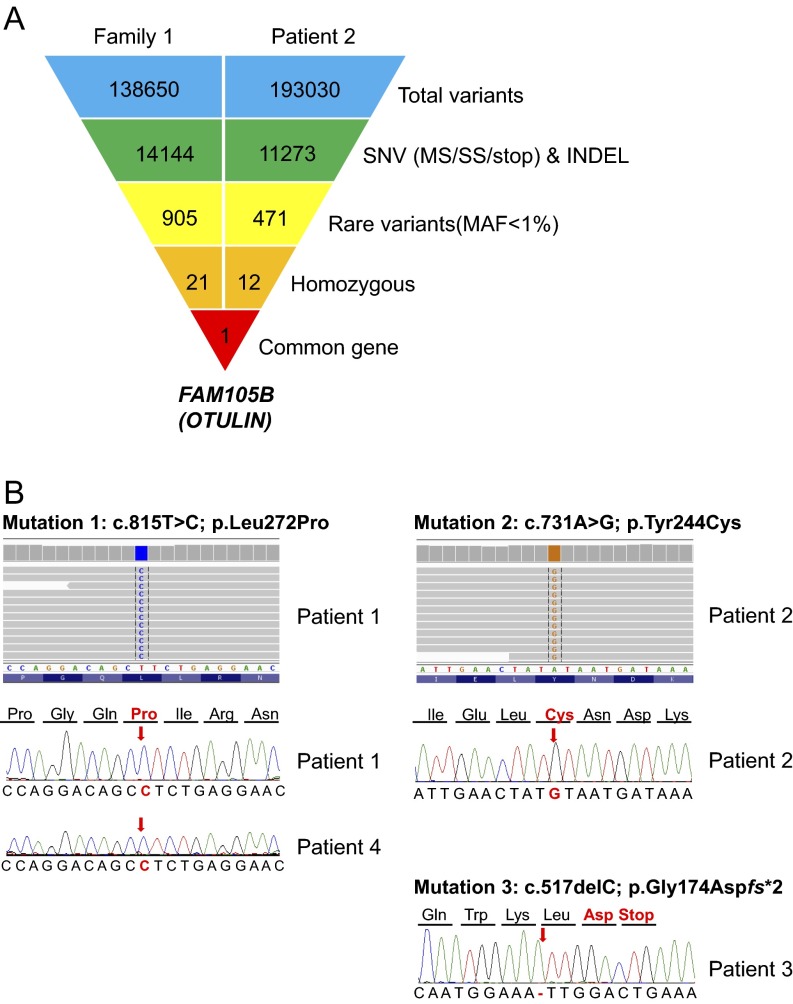
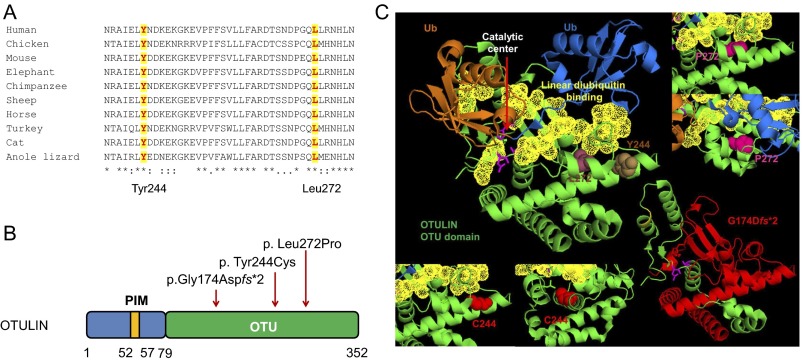
Systemic autoinflammatory diseases are caused by mutations in genes that function in innate immunity. Here, we report an autoinflammatory disease caused by loss-of-function mutations in OTULIN (FAM105B), encoding a deubiquitinase with linear linkage specificity. We identified two missense and one frameshift mutations in one Pakistani and two Turkish families with four affected patients. Patients presented with neonatal-onset fever, neutrophilic dermatitis/panniculitis, and failure to thrive, but without obvious primary immunodeficiency. HEK293 cells transfected with mutated OTULIN had decreased enzyme activity relative to cells transfected with WT OTULIN, and showed a substantial defect in the linear deubiquitination of target molecules. Stimulated patients' fibroblasts and peripheral blood mononuclear cells showed evidence for increased signaling in the canonical NF-κB pathway and accumulated linear ubiquitin aggregates. Levels of proinflammatory cytokines were significantly increased in the supernatants of stimulated primary cells and serum samples. This discovery adds to the emerging spectrum of human diseases caused by defects in the ubiquitin pathway and suggests a role for targeted cytokine therapies.
Keywords: NF-κB pathway; OTULIN; autoinflammatory disease; cytokines; linear deubiquitinase.
Conflict of interest statement
Conflict of interest statement: S.Ö. received royalties for consulting and speaking from Novartis and SOBI. All other authors declare no conflict of interest.
Figures
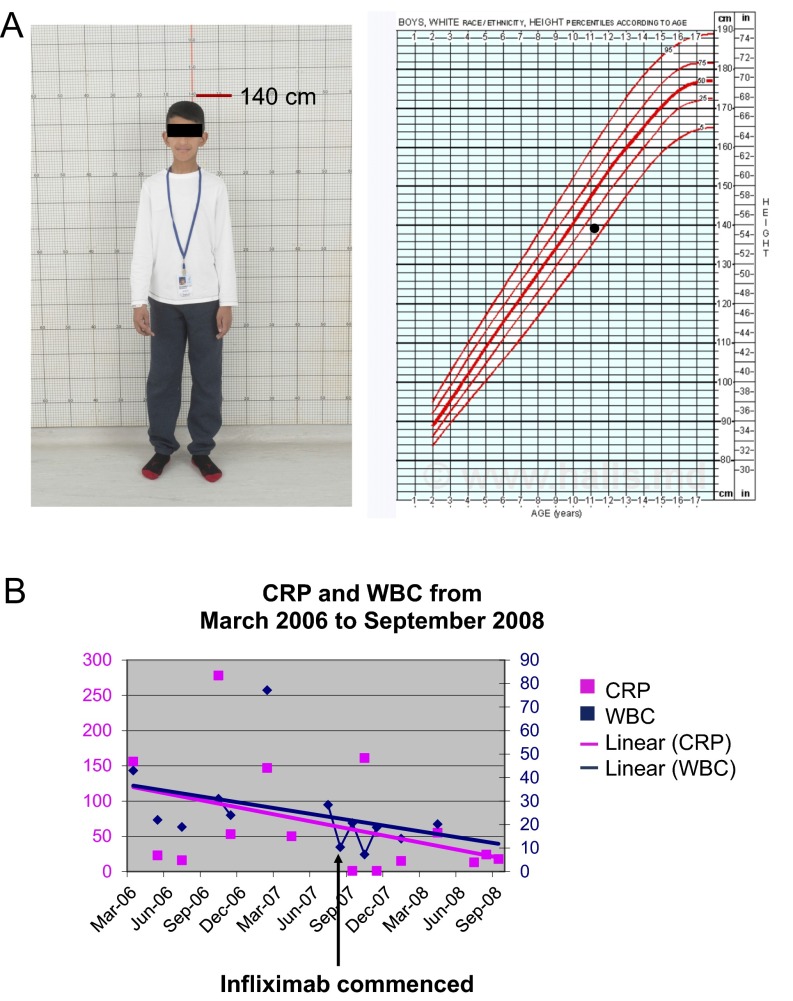
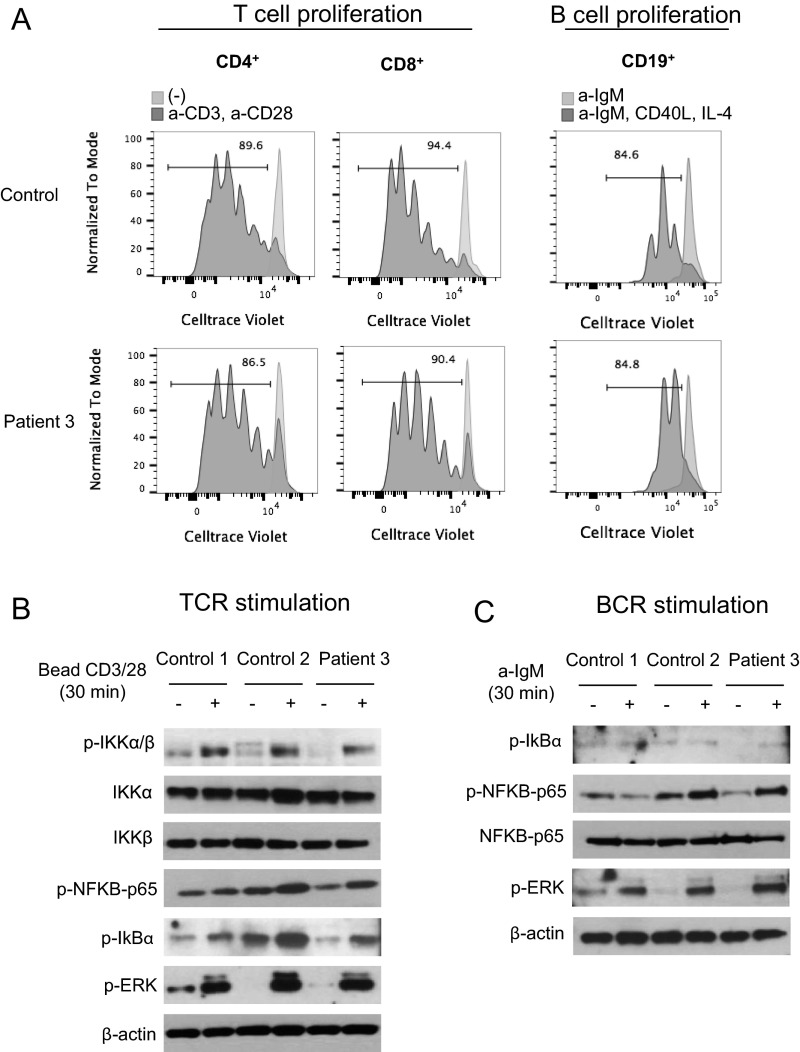
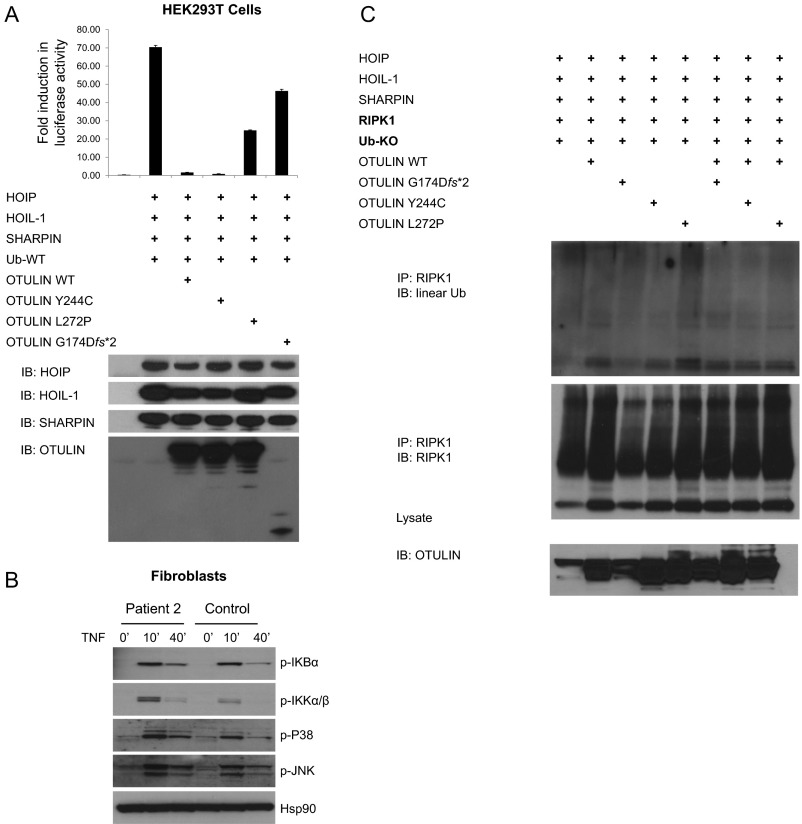
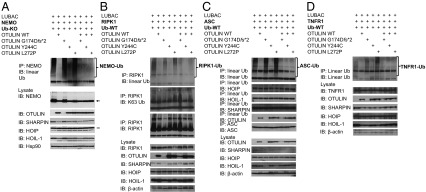
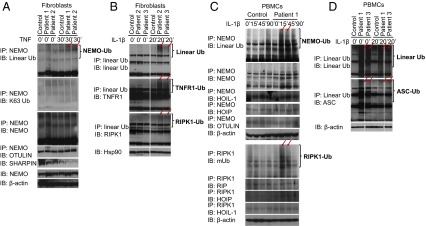
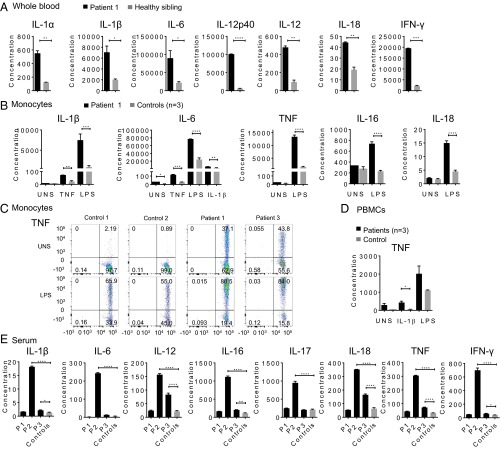
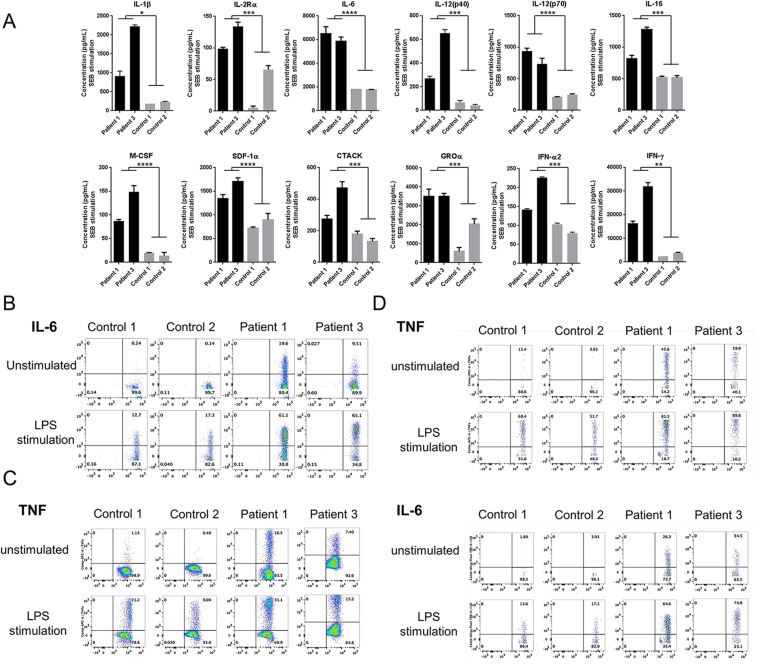
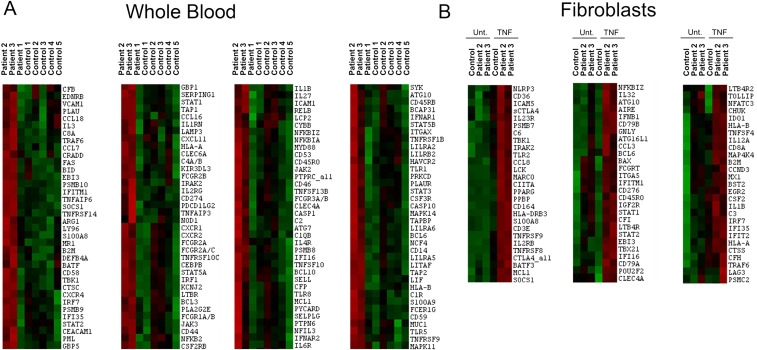
References
-
- Gerlach B, et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature. 2011;471(7340):591–596. - PubMed
-
- Tokunaga F, et al. SHARPIN is a component of the NF-κB-activating linear ubiquitin chain assembly complex. Nature. 2011;471(7340):633–636. - PubMed
-
- Ombrello MJ, Kastner DL, Milner JD. HOIL and water: The two faces of HOIL-1 deficiency. Nat Immunol. 2012;13(12):1133–1135. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
