

Assembly-based inference of B-cell receptor repertoires from short read RNA sequencing data with V'DJer
- PMID: 27559159
- PMCID: PMC5167060
- DOI: 10.1093/bioinformatics/btw526
Assembly-based inference of B-cell receptor repertoires from short read RNA sequencing data with V'DJer
Abstract
Motivation: B-cell receptor (BCR) repertoire profiling is an important tool for understanding the biology of diverse immunologic processes. Current methods for analyzing adaptive immune receptor repertoires depend upon PCR amplification of VDJ rearrangements followed by long read amplicon sequencing spanning the VDJ junctions. While this approach has proven to be effective, it is frequently not feasible due to cost or limited sample material. Additionally, there are many existing datasets where short-read RNA sequencing data are available but PCR amplified BCR data are not.
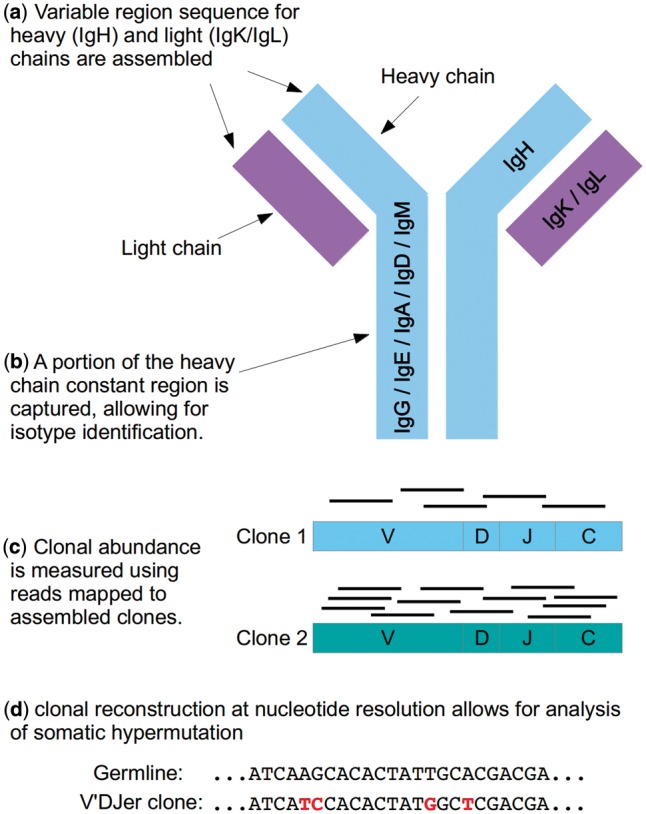
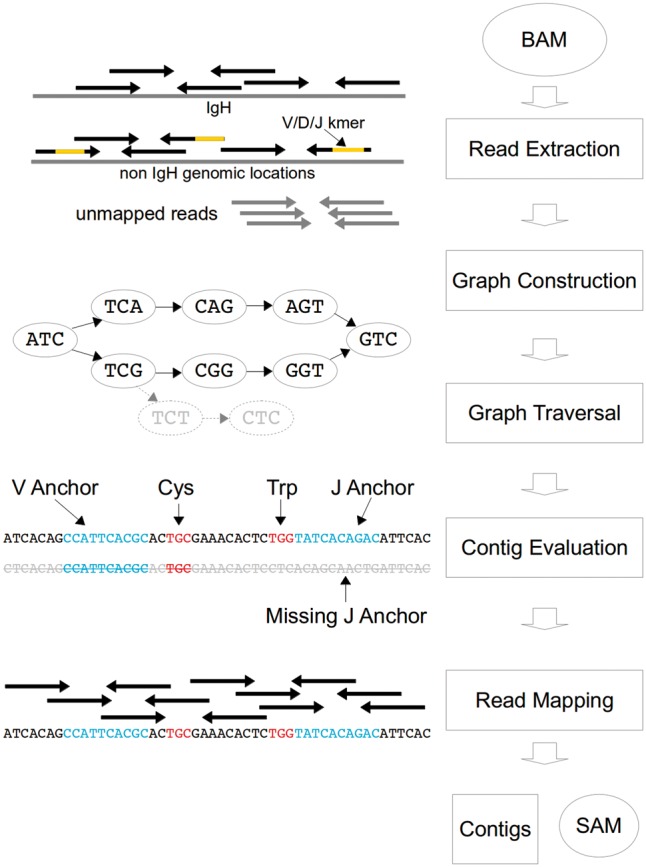
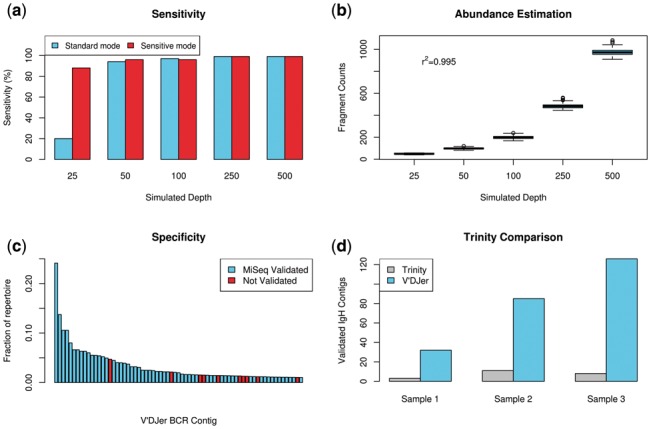
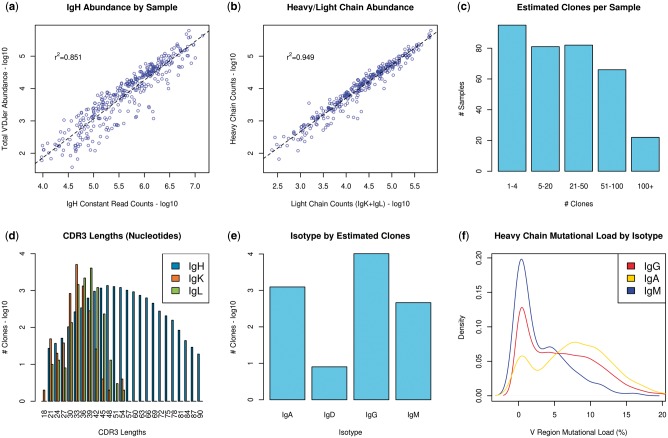
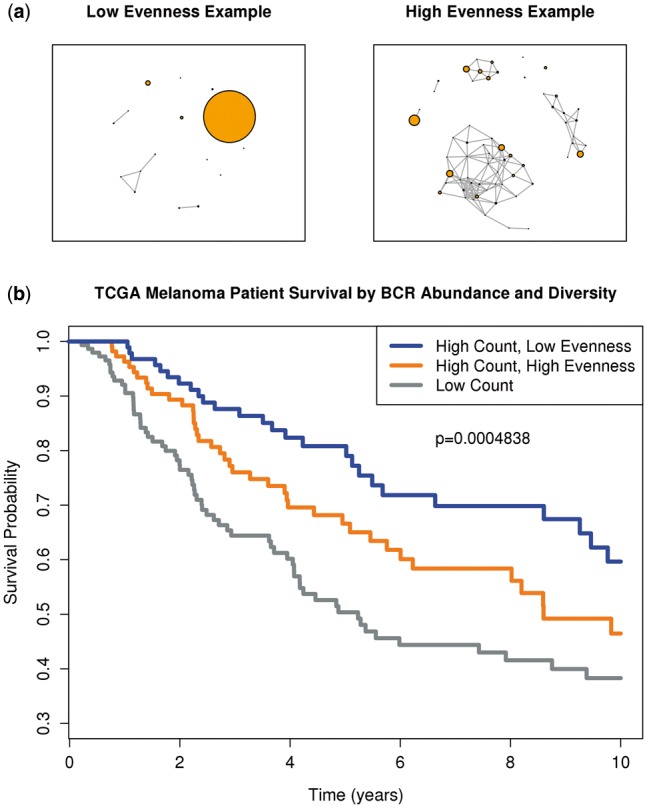
Results: We present here V'DJer, an assembly-based method that reconstructs adaptive immune receptor repertoires from short-read RNA sequencing data. This method captures expressed BCR loci from a standard RNA-seq assay. We applied this method to 473 Melanoma samples from The Cancer Genome Atlas and demonstrate V'DJer's ability to accurately reconstruct BCR repertoires from short read mRNA-seq data.
Availability and implementation: V'DJer is implemented in C/C ++, freely available for academic use and can be downloaded from Github: https://github.com/mozack/vdjer CONTACT: benjamin_vincent@med.unc.edu or parkerjs@email.unc.eduSupplementary information: Supplementary data are available at Bioinformatics online.
© The Author 2016. Published by Oxford University Press.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
