

Prediction of Protein Structure Using Surface Accessibility Data
- PMID: 27560616
- PMCID: PMC5026166
- DOI: 10.1002/anie.201604788
Prediction of Protein Structure Using Surface Accessibility Data
Abstract
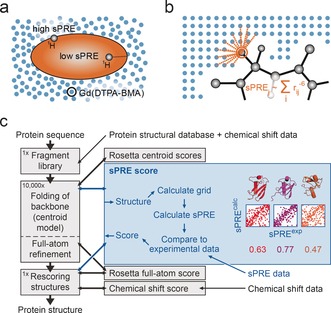
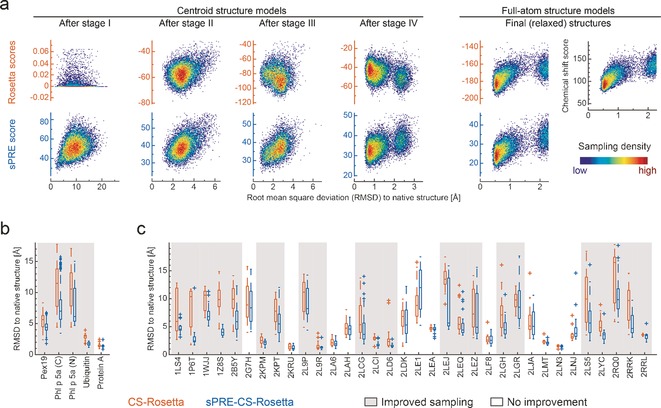
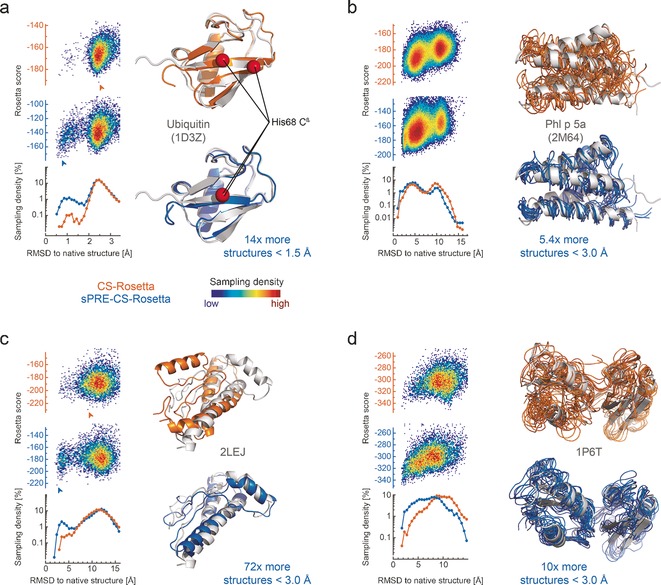
An approach to the de novo structure prediction of proteins is described that relies on surface accessibility data from NMR paramagnetic relaxation enhancements by a soluble paramagnetic compound (sPRE). This method exploits the distance-to-surface information encoded in the sPRE data in the chemical shift-based CS-Rosetta de novo structure prediction framework to generate reliable structural models. For several proteins, it is demonstrated that surface accessibility data is an excellent measure of the correct protein fold in the early stages of the computational folding algorithm and significantly improves accuracy and convergence of the standard Rosetta structure prediction approach.
Keywords: CS-Rosetta; NMR spectroscopy; paramagnetic relaxation; protein structure prediction; structural biology.
© 2016 The Authors. Published by Wiley-VCH Verlag GmbH & Co. KGaA.
Figures
Similar articles
-
Recent Advances in NMR Protein Structure Prediction with ROSETTA.Int J Mol Sci. 2023 Apr 25;24(9):7835. doi: 10.3390/ijms24097835. Int J Mol Sci. 2023. PMID: 37175539 Free PMC article. Review.
-
Protein structure prediction using sparse NOE and RDC restraints with Rosetta in CASP13.Proteins. 2019 Dec;87(12):1341-1350. doi: 10.1002/prot.25769. Epub 2019 Jul 18. Proteins. 2019. PMID: 31292988 Free PMC article.
-
De novo protein structure generation from incomplete chemical shift assignments.J Biomol NMR. 2009 Feb;43(2):63-78. doi: 10.1007/s10858-008-9288-5. Epub 2008 Nov 26. J Biomol NMR. 2009. PMID: 19034676 Free PMC article.
-
3D Computational Modeling of Proteins Using Sparse Paramagnetic NMR Data.Methods Mol Biol. 2017;1526:3-21. doi: 10.1007/978-1-4939-6613-4_1. Methods Mol Biol. 2017. PMID: 27896733
-
Solvent paramagnetic relaxation enhancement as a versatile method for studying structure and dynamics of biomolecular systems.Prog Nucl Magn Reson Spectrosc. 2022 Oct-Dec;132-133:113-139. doi: 10.1016/j.pnmrs.2022.09.001. Epub 2022 Sep 21. Prog Nucl Magn Reson Spectrosc. 2022. PMID: 36496256 Review.
Cited by
-
Characterization of Protein-Protein Interfaces in Large Complexes by Solid-State NMR Solvent Paramagnetic Relaxation Enhancements.J Am Chem Soc. 2017 Sep 6;139(35):12165-12174. doi: 10.1021/jacs.7b03875. Epub 2017 Aug 25. J Am Chem Soc. 2017. PMID: 28780861 Free PMC article.
-
AssignSLP_GUI, a software tool exploiting AI for NMR resonance assignment of sparsely labeled proteins.J Magn Reson. 2022 Dec;345:107336. doi: 10.1016/j.jmr.2022.107336. Epub 2022 Nov 19. J Magn Reson. 2022. PMID: 36442299 Free PMC article.
-
Amino Acid Insertion Frequencies Arising from Photoproducts Generated Using Aliphatic Diazirines.J Am Soc Mass Spectrom. 2017 Oct;28(10):2011-2021. doi: 10.1007/s13361-017-1730-z. Epub 2017 Aug 10. J Am Soc Mass Spectrom. 2017. PMID: 28799075
-
A cation-π interaction in a transmembrane helix of vacuolar ATPase retains the proton-transporting arginine in a hydrophobic environment.J Biol Chem. 2018 Dec 7;293(49):18977-18988. doi: 10.1074/jbc.RA118.005276. Epub 2018 Sep 12. J Biol Chem. 2018. PMID: 30209131 Free PMC article.
-
Utilization of Hydrophobic Microenvironment Sensitivity in Diethylpyrocarbonate Labeling for Protein Structure Prediction.Anal Chem. 2021 Jun 15;93(23):8188-8195. doi: 10.1021/acs.analchem.1c00395. Epub 2021 Jun 1. Anal Chem. 2021. PMID: 34061512 Free PMC article.
References
-
- None
-
- Göbl C., Madl T., Simon B., Sattler M., Prog. Nucl. Magn. Reson. Spectrosc. 2014, 80, 26–63; - PubMed
-
- Cavanagh J., Protein NMR Spectroscopy: Principles and Practice , 2nd ed., Academic Press, Amsterdam, Boston, 2007;
-
- Rule G. S., Hitchens T. K., Fundamentals of Protein NMR Spectroscopy, Springer, Dordrecht, 2006.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
