

Cortisol-induced immune suppression by a blockade of lymphocyte egress in traumatic brain injury
- PMID: 27561600
- PMCID: PMC5000452
- DOI: 10.1186/s12974-016-0663-y
Cortisol-induced immune suppression by a blockade of lymphocyte egress in traumatic brain injury
Abstract
Background: Acute traumatic brain injury (TBI) represents one of major causes of mortality and disability in the USA. Neuroinflammation has been regarded both beneficial and detrimental, probably in a time-dependent fashion.
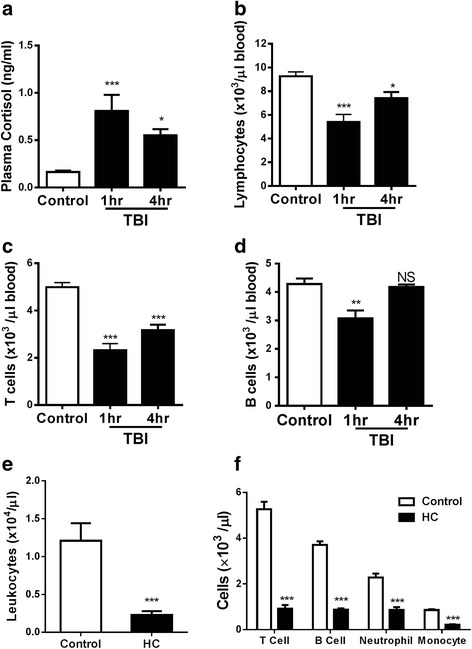
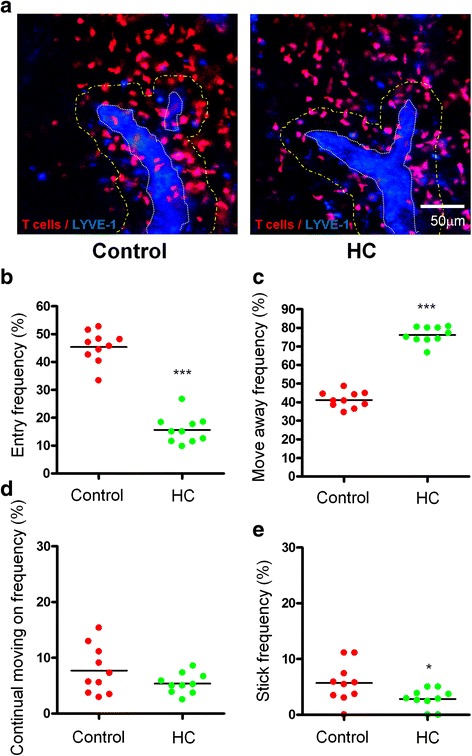
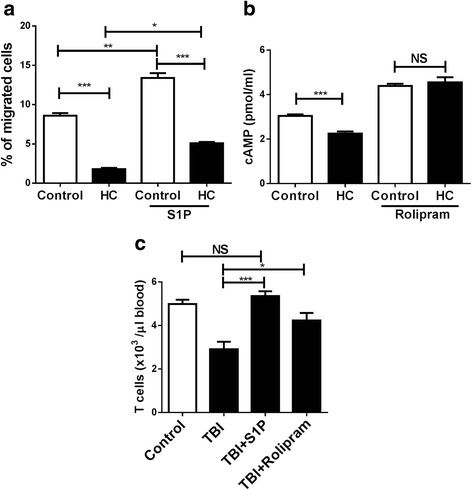
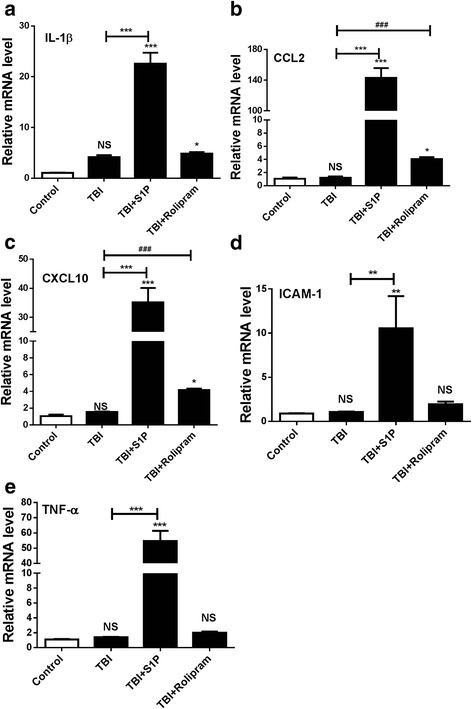
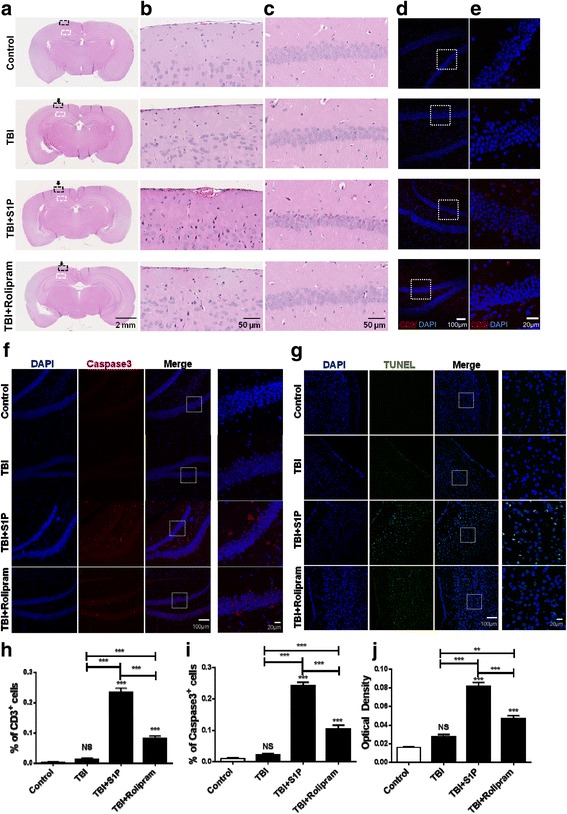
Methods: To address a role for neuroinflammation in brain injury, C57BL/6 mice were subjected to a closed head mild TBI (mTBI) by a standard controlled cortical impact, along with or without treatment of sphingosine 1-phosphate (S1P) or rolipram, after which the brain tissue of the impact site was evaluated for cell morphology via histology, inflammation by qRT-PCR and T cell staining, and cell death with Caspase-3 and TUNEL staining. Circulating lymphocytes were quantified by flow cytometry, and plasma hydrocortisone was analyzed by LC-MS/MS. To investigate the mechanism whereby cortisol lowered the number of peripheral T cells, T cell egress was tracked in lymph nodes by intravital confocal microscopy after hydrocortisone administration.
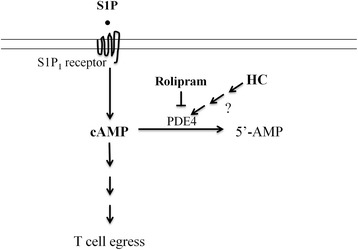
Results: We detected a decreased number of circulating lymphocytes, in particular, T cells soon after mTBI, which was inversely correlated with a transient and robust increase of plasma cortisol. The transient lymphocytopenia might be caused by cortisol in part via a blockade of lymphocyte egress as demonstrated by the ability of cortisol to inhibit T cell egress from the secondary lymphoid tissues. Moreover, exogenous hydrocortisone severely suppressed periphery lymphocytes in uninjured mice, whereas administering an egress-promoting agent S1P normalized circulating T cells in mTBI mice and increased T cells in the injured brain. Likewise, rolipram, a cAMP phosphodiesterase inhibitor, was also able to elevate cAMP levels in T cells in the presence of hydrocortisone in vitro and abrogate the action of cortisol in mTBI mice. The investigation demonstrated that the number of circulating T cells in the early phase of TBI was positively correlated with T cell infiltration and inflammatory responses as well as cell death at the cerebral cortex and hippocampus beneath the impact site.
Conclusions: Decreases in intracellular cAMP might be part of the mechanism behind cortisol-mediated blockade of T cell egress. The study argues strongly for a protective role of cortisol-induced immune suppression in the early stage of TBI.
Keywords: Cortisol; Inflammation; T lymphocytes; TBI; cAMP.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
