

doi: 10.1038/srep31232.
Metadynamic metainference: Enhanced sampling of the metainference ensemble using metadynamics
Affiliations
- PMID: 27561930
- PMCID: PMC4999896
- DOI: 10.1038/srep31232
Item in Clipboard
Metadynamic metainference: Enhanced sampling of the metainference ensemble using metadynamics
Sci Rep.
.
Abstract
Accurate and precise structural ensembles of proteins and macromolecular complexes can be obtained with metainference, a recently proposed Bayesian inference method that integrates experimental information with prior knowledge and deals with all sources of errors in the data as well as with sample heterogeneity. The study of complex macromolecular systems, however, requires an extensive conformational sampling, which represents a separate challenge. To address such challenge and to exhaustively and efficiently generate structural ensembles we combine metainference with metadynamics and illustrate its application to the calculation of the free energy landscape of the alanine dipeptide.
Figures
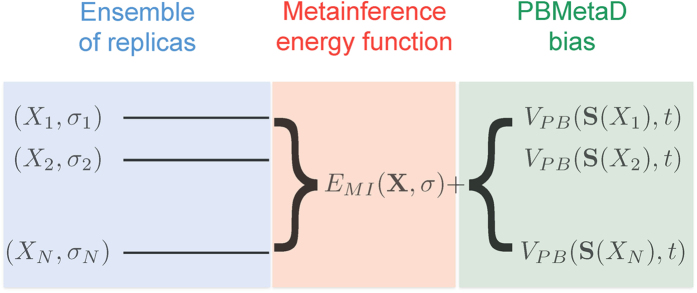
In M&M, an ensemble of models is simulated in parallel. Typically a model is composed by the following variables: the coordinates of the system X and a list of variables σ that represent the level of noise in the data. In principle one can associate a σ variable to each experimental data point (Eq. 19), or use an outlier model of noise that requires a single σ for all data points (Eq. 20). The replicas are coupled by the metainference energy function, which is composed by different terms: one that describes prior information on the system X (for example a molecular mechanics force field), one that describes prior information on the σ variable (typically a Jeffrey’s prior), and one term that describes the agreement of the models with the experimental data. This last energy term couples the multiple replicas, as experimental data are expected to be generated by the average over an ensemble of conformations. To accelerate sampling, M&M adds to each replica the bias potential of PBMetaD. This is defined as a function of multiple CVs, which are selected from the user based on a priori knowledge of the system to include all the slow modes that need to be accelerated. The PBMetaD bias potential is defined in terms of multiple potentials (one for each CVs), which are stored on a common grid and shared among all replicas. In doing so, the multiple replicas all contribute to fill in parallel the underlying free-energy landascapes and thus to accelerate sampling of the entire ensemble.
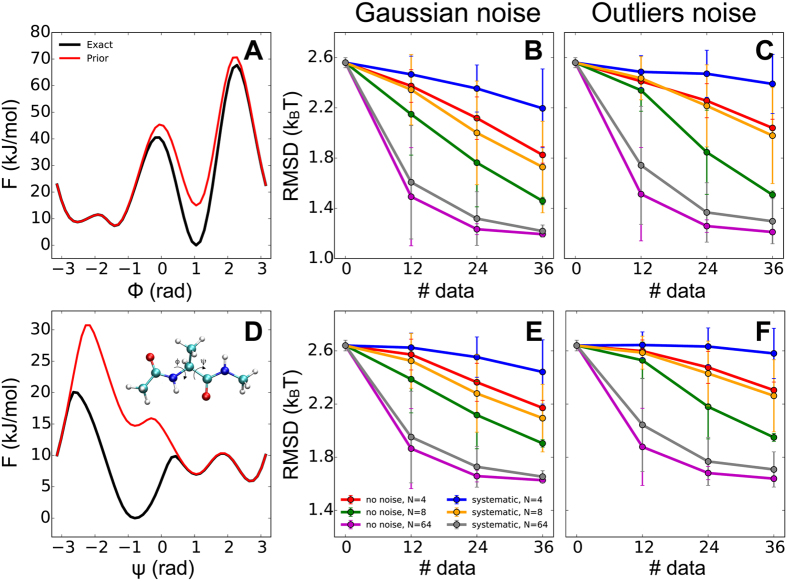
We assumed that the ensemble generated by the prior alone (AMBER99SB-ILDN force field, (A,D) red lines) is inaccurate, and that in the actual ensemble the local minimum Cax is more populated (A,D, black lines). We then used M&M to combine the inaccurate prior with synthetic experimental data generated as averages on the correct ensemble. We calculated the error in the reconstructed free energies as a function of the backbone dihedrals ϕ (upper panels) and ψ (lower panels), using Gaussian (B,E) and outliers noise models (C,F). For both noise models, we benchmarked the method as a function of the number of data points, the level of noise in the data, and the number of replicas used.
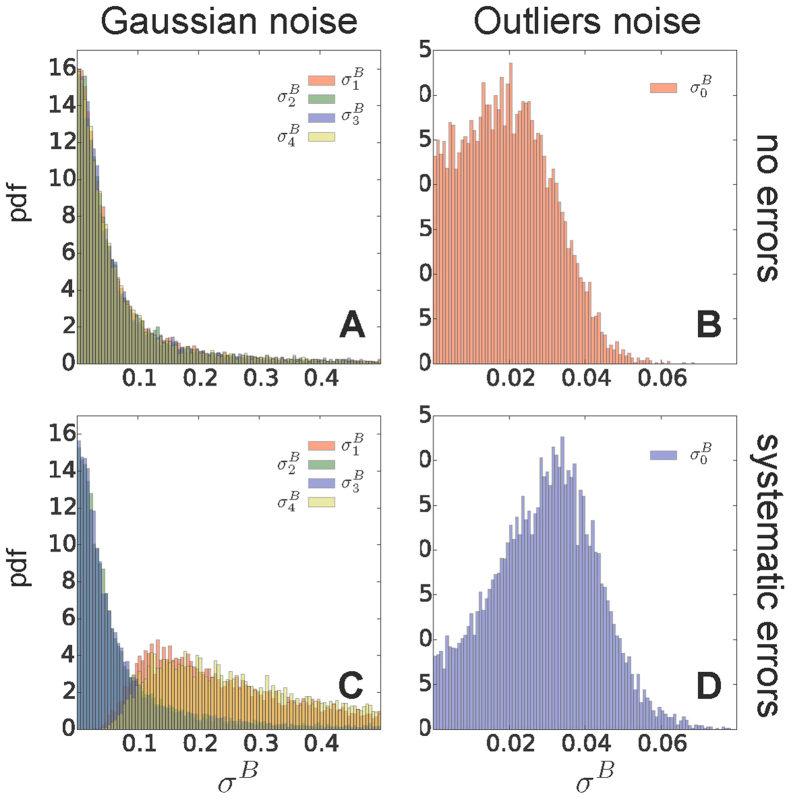
The distributions of the σB parameter in representative runs with 8 replicas and all the 36 data points available, using the Gaussian (AC) and outliers noise models (BD), in absence (top panels) and presence of systematic errors (bottom panels). When using Gaussian noise, we plot the distributions of the uncertainty parameter associated to four representative data points ( ). When systematic errors are present, we selected two outliers (
). When systematic errors are present, we selected two outliers ( and
and  ) and two data points not affected by error (
) and two data points not affected by error ( and
and  ). When using the outliers model, we plot the distribution of the typical dataset uncertainty
). When using the outliers model, we plot the distribution of the typical dataset uncertainty  .
.
). When systematic errors are present, we selected two outliers ( and ) and two data points not affected by error ( and ). When using the outliers model, we plot the distribution of the typical dataset uncertainty .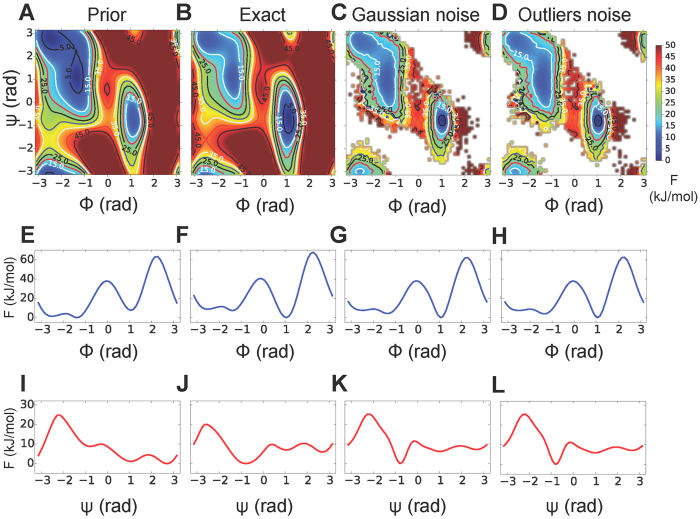
Free energy of alanine dipeptide as a function of the backbone dihedrals obtained with the AMBER99SB-ILDN prior alone (A) and with the correction to lower the free energy of the local minimum Cax (B). The latter is considered our reference (exact) free energy. Free energy obtained from reweighting a M&M simulation carried out using 64 replicas, all the 36 data points available (without addition of systematic errors), the Gaussian noise model with one uncertainty parameter for each data point (C) and the outliers noise model with one parameter per dataset (D). The visualization is truncated at 50 kJ/mol from the global minimum. For each case (prior, exact, Gaussian and outliers noise), we also reported the monodimensional free energies as a function of the dihedrals ϕ (E–H) and ψ (I–L), calculated directly from the bias potentials. The same analysis using a dataset with systematic errors is reported in Fig. S7.
Similar articles
-
A Practical Guide to the Simultaneous Determination of Protein Structure and Dynamics Using Metainference.Methods Mol Biol. 2019;2022:313-340. doi: 10.1007/978-1-4939-9608-7_13. Methods Mol Biol. 2019. PMID: 31396909
-
Metadynamic metainference: Convergence towards force field independent structural ensembles of a disordered peptide.J Chem Phys. 2017 Apr 28;146(16):165102. doi: 10.1063/1.4981211. J Chem Phys. 2017. PMID: 28456189
-
Guide for determination of protein structural ensembles by combining cryo-EM data with metadynamics.FEBS Open Bio. 2023 Jul;13(7):1193-1203. doi: 10.1002/2211-5463.13542. Epub 2023 Jan 9. FEBS Open Bio. 2023. PMID: 36562694 Free PMC article.
-
Using simulation to interpret experimental data in terms of protein conformational ensembles.Curr Opin Struct Biol. 2017 Apr;43:79-87. doi: 10.1016/j.sbi.2016.11.018. Epub 2016 Dec 9. Curr Opin Struct Biol. 2017. PMID: 27940377 Review.
-
Advances in uncovering the mechanisms of macromolecular conformational entropy.Nat Chem Biol. 2025 May;21(5):623-634. doi: 10.1038/s41589-025-01879-3. Epub 2025 Apr 24. Nat Chem Biol. 2025. PMID: 40275100 Free PMC article. Review.
Cited by
-
Determination of the Structure and Dynamics of the Fuzzy Coat of an Amyloid Fibril of IAPP Using Cryo-Electron Microscopy.Biochemistry. 2023 Aug 15;62(16):2407-2416. doi: 10.1021/acs.biochem.3c00010. Epub 2023 Jul 21. Biochemistry. 2023. PMID: 37477459 Free PMC article.
-
A conformational fingerprint for amyloidogenic light chains.Elife. 2025 Mar 3;13:RP102002. doi: 10.7554/eLife.102002. Elife. 2025. PMID: 40028903 Free PMC article.
-
Refinement of α-Synuclein Ensembles Against SAXS Data: Comparison of Force Fields and Methods.Front Mol Biosci. 2021 Apr 22;8:654333. doi: 10.3389/fmolb.2021.654333. eCollection 2021. Front Mol Biosci. 2021. PMID: 33968988 Free PMC article.
-
Reweighting of molecular simulations with explicit-solvent SAXS restraints elucidates ion-dependent RNA ensembles.Nucleic Acids Res. 2021 Aug 20;49(14):e84. doi: 10.1093/nar/gkab459. Nucleic Acids Res. 2021. PMID: 34107023 Free PMC article.
-
Methods of probing the interactions between small molecules and disordered proteins.Cell Mol Life Sci. 2017 Sep;74(17):3225-3243. doi: 10.1007/s00018-017-2563-4. Epub 2017 Jun 19. Cell Mol Life Sci. 2017. PMID: 28631009 Free PMC article. Review.
References
-
- Rieping W., Habeck M. & Nilges M. Inferential structure determination. Science 309, 303–306 (2005). - PubMed
-
- Cavalli A., Camilloni C. & Vendruscolo M. Molecular dynamics simulations with replica-averaged structural restraints generate structural ensembles according to the maximum entropy principle. J. Chem. Phys. 138, 094112 (2013). - PubMed
-
- Lindorff-Larsen K., Best R. B., Depristo M. A., Dobson C. M. & Vendruscolo M. Simultaneous determination of protein structure and dynamics. Nature 433, 128–132 (2005). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
