

Podocyte NF-κB is dispensable for the pathogenesis of renal ischemia-reperfusion injury
- PMID: 27565904
- PMCID: PMC5002916
- DOI: 10.14814/phy2.12912
Podocyte NF-κB is dispensable for the pathogenesis of renal ischemia-reperfusion injury
Abstract
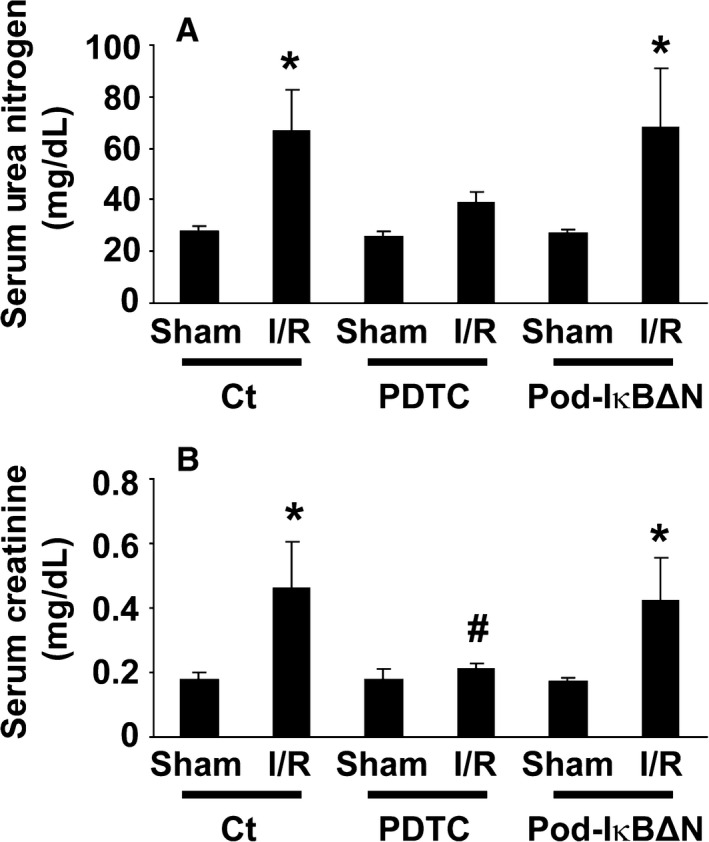
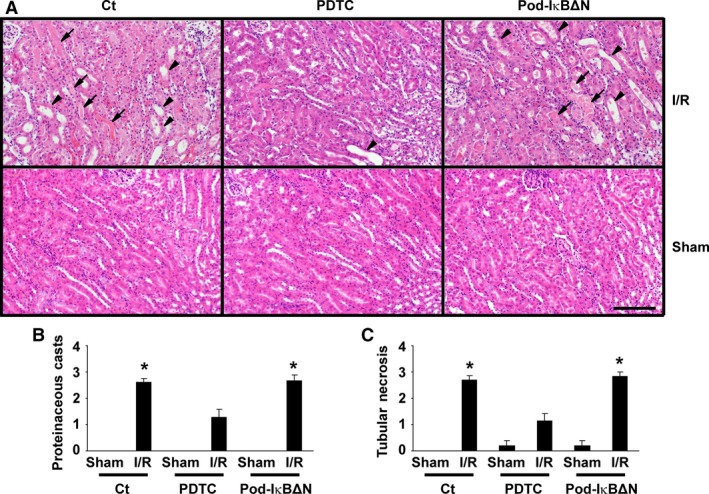
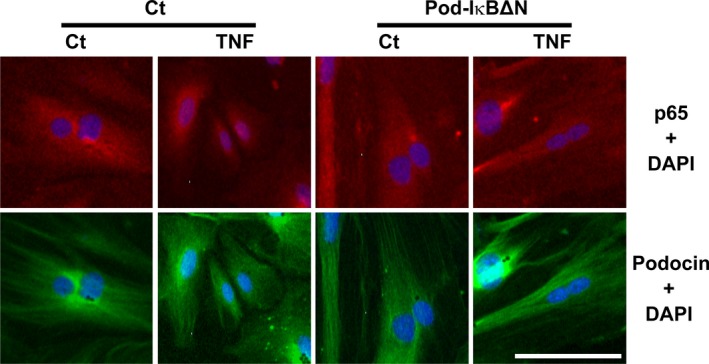
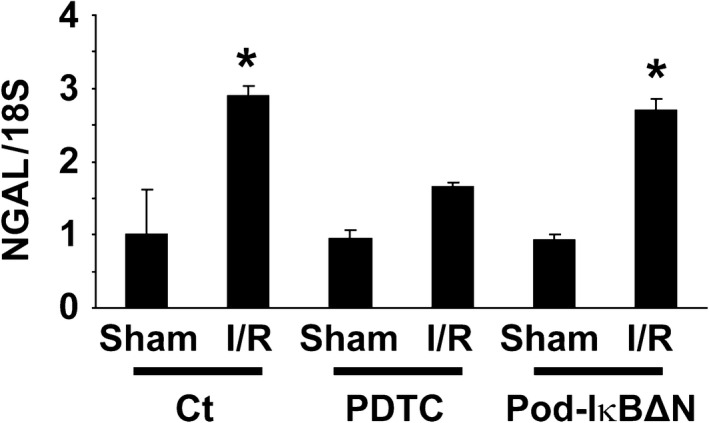
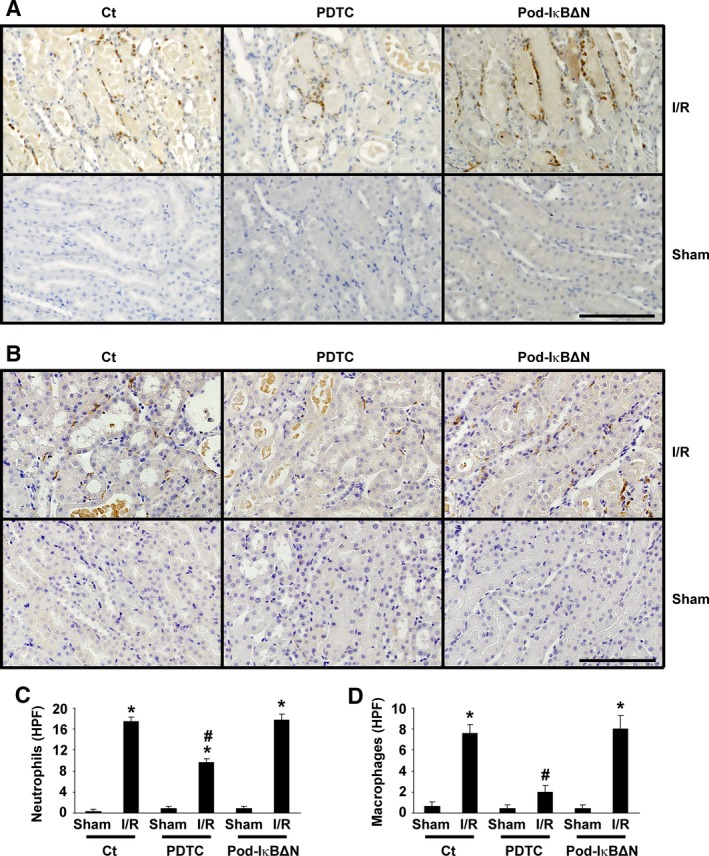
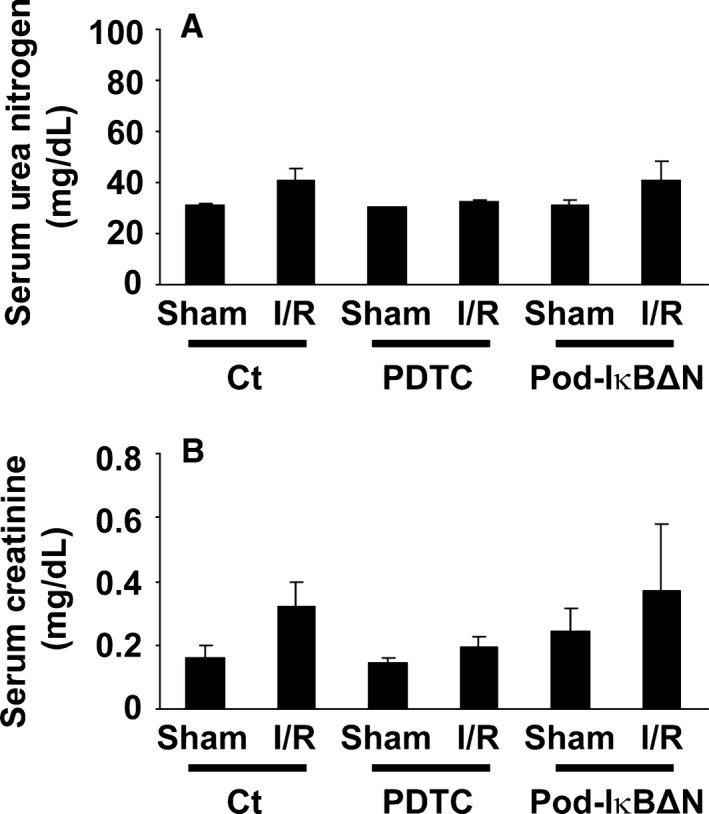
Podocytes play a central role in the formation of the glomerular filtration barrier in the kidney, and their dysfunction has been shown to result in multiple proteinuric kidney diseases. In this study, we sought to determine whether NF-κB, a proinflammatory signaling, within podocytes was involved in renal ischemia-reperfusion (I/R) injury. Podocyte-specific IκBΔN transgenic (Pod-IκBΔN) mice, in which NF-κB was inhibited specifically in podocytes, were generated by the Cre-loxP technology, and their phenotype was compared with control mice after bilateral renal ischemia. The effect of systemic administration of a NF-κB inhibitor, pyrrolidinedithiocarbamate (PDTC), on renal I/R injury was also examined. Pod-IκBΔN mice were phenotypically normal before surgery. Following renal I/R injury, serum concentrations of urea nitrogen and creatinine were elevated in both Pod-IκBΔN and control mice to a similar extent, whereas PDTC treatment attenuated the elevation of these parameters. Renal histological damage in I/R-injured Pod-IκBΔN mice was also similar to I/R-injured control mice, although it was improved by PDTC treatment. Moreover, I/R induced accumulation of inflammatory cells, such as neutrophils and macrophages, was reduced by PDTC treatment, but not by podocyte-specific NF-κB inhibition. These results provide evidence that the NF-κB activity in podocytes does not contribute to the pathogenesis of renal I/R injury.
Keywords: Acute kidney injury; NF‐κB; ischemia‐reperfusion; podocytes.
© 2016 The Authors. Physiological Reports published by Wiley Periodicals, Inc. on behalf of the American Physiological Society and The Physiological Society.
Figures
Similar articles
-
Podocyte-specific NF-κB inhibition ameliorates proteinuria in adriamycin-induced nephropathy in mice.Clin Exp Nephrol. 2017 Feb;21(1):16-26. doi: 10.1007/s10157-016-1268-6. Epub 2016 Apr 18. Clin Exp Nephrol. 2017. PMID: 27089875
-
Nuclear factor-kappaB inhibition by pyrrolidinedithiocarbamate attenuates gastric ischemia-reperfusion injury in rats.Can J Physiol Pharmacol. 2005 Jun;83(6):483-92. doi: 10.1139/y05-034. Can J Physiol Pharmacol. 2005. PMID: 16049548
-
NF-κB signaling pathway-enhanced complement activation mediates renal injury in trichloroethylene-sensitized mice.J Immunotoxicol. 2018 Dec;15(1):63-72. doi: 10.1080/1547691X.2017.1420712. J Immunotoxicol. 2018. PMID: 29534626 Free PMC article.
-
Protective effect of pyrrolidine dithiocarbamate on liver injury induced by intestinal ischemia-reperfusion in rats.Hepatobiliary Pancreat Dis Int. 2006 Feb;5(1):90-5. Hepatobiliary Pancreat Dis Int. 2006. PMID: 16481291
-
Crocin Protects Podocytes Against Oxidative Stress and Inflammation Induced by High Glucose Through Inhibition of NF-κB.Cell Physiol Biochem. 2017;42(4):1481-1492. doi: 10.1159/000479212. Epub 2017 Jul 18. Cell Physiol Biochem. 2017. PMID: 28719912
Cited by
-
Recent Approaches to Targeting Canonical NFκB Signaling in the Early Inflammatory Response to Renal IRI.J Am Soc Nephrol. 2021 Sep;32(9):2117-2124. doi: 10.1681/ASN.2021010069. Epub 2021 Jun 9. J Am Soc Nephrol. 2021. PMID: 34108233 Free PMC article. Review.
-
Foxa2 attenuates steatosis and inhibits the NF-κB/IKK signaling pathway in nonalcoholic fatty liver disease.PeerJ. 2023 Dec 7;11:e16466. doi: 10.7717/peerj.16466. eCollection 2023. PeerJ. 2023. PMID: 38084145 Free PMC article.
-
NFκB and Kidney Injury.Front Immunol. 2019 Apr 16;10:815. doi: 10.3389/fimmu.2019.00815. eCollection 2019. Front Immunol. 2019. PMID: 31040851 Free PMC article. Review.
-
Maresin 1 mitigates renal ischemia/reperfusion injury in mice via inhibition of the TLR4/MAPK/NF-κB pathways and activation of the Nrf2 pathway.Drug Des Devel Ther. 2019 Feb 20;13:739-745. doi: 10.2147/DDDT.S188654. eCollection 2019. Drug Des Devel Ther. 2019. PMID: 30863013 Free PMC article.
References
-
- Asano, T. , Niimura F., Pastan I., Fogo A. B., Ichikawa I., and Matsusaka T.. 2005. Permanent genetic tagging of podocytes: fate of injured podocytes in a mouse model of glomerular sclerosis. J. Am. Soc. Nephrol. 16:2257–2262. - PubMed
-
- Brähler, S. , Ising C., Hagmann H., Rasmus M., Hoehne M., Kurschat C., et al. 2012. Intrinsic proinflammatory signaling in podocytes contributes to podocyte damage and prolonged proteinuria. Am. J. Physiol. Renal. Physiol. 303:F1473–F1485. - PubMed
-
- Brown, K. , Gerstberger S., Carlson L., Franzoso G., and Siebenlist U.. 1995. Control of IκB‐α proteolysis by site‐specific, signal‐induced phosphorylation. Science 267:1485–1488. - PubMed
-
- Cao, C. C. , Ding X. Q., Ou Z. L., Liu C. F., Li P., Wang L., et al. 2004. In vivo transfection of NF‐κB decoy oligonucleotides attenuate renal ischemia/reperfusion injury in rats. Kidney Int. 65:834–845. - PubMed
-
- Donnahoo, K. K. , Meldrum D. R., Shenkar R., Chung C. S., Abraham E., and Harken A. H.. 2000. Early renal ischemia, with or without reperfusion, activates NFκB and increases TNF‐α bioactivity in the kidney. J. Urol. 163:1328–1332. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
