

Molecular and structural considerations of TF-DNA binding for the generation of biologically meaningful and accurate phylogenetic footprinting analysis: the LysR-type transcriptional regulator family as a study model
- PMID: 27567672
- PMCID: PMC5002191
- DOI: 10.1186/s12864-016-3025-3
Molecular and structural considerations of TF-DNA binding for the generation of biologically meaningful and accurate phylogenetic footprinting analysis: the LysR-type transcriptional regulator family as a study model
Abstract
Background: The goal of most programs developed to find transcription factor binding sites (TFBSs) is the identification of discrete sequence motifs that are significantly over-represented in a given set of sequences where a transcription factor (TF) is expected to bind. These programs assume that the nucleotide conservation of a specific motif is indicative of a selective pressure required for the recognition of a TF for its corresponding TFBS. Despite their extensive use, the accuracies reached with these programs remain low. In many cases, true TFBSs are excluded from the identification process, especially when they correspond to low-affinity but important binding sites of regulatory systems.
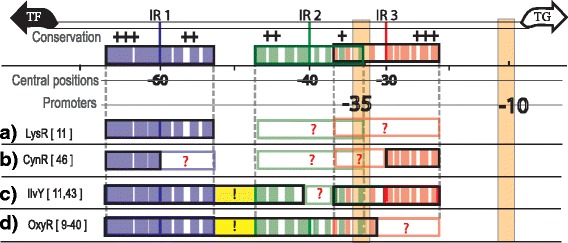
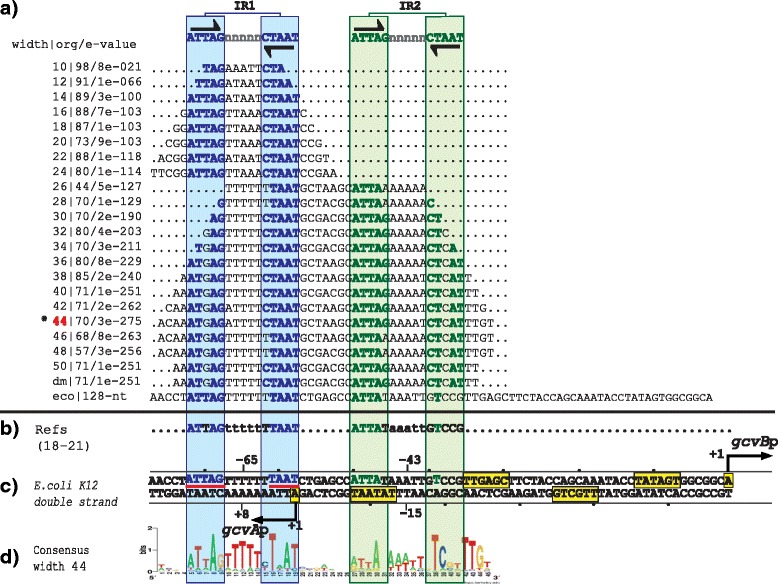
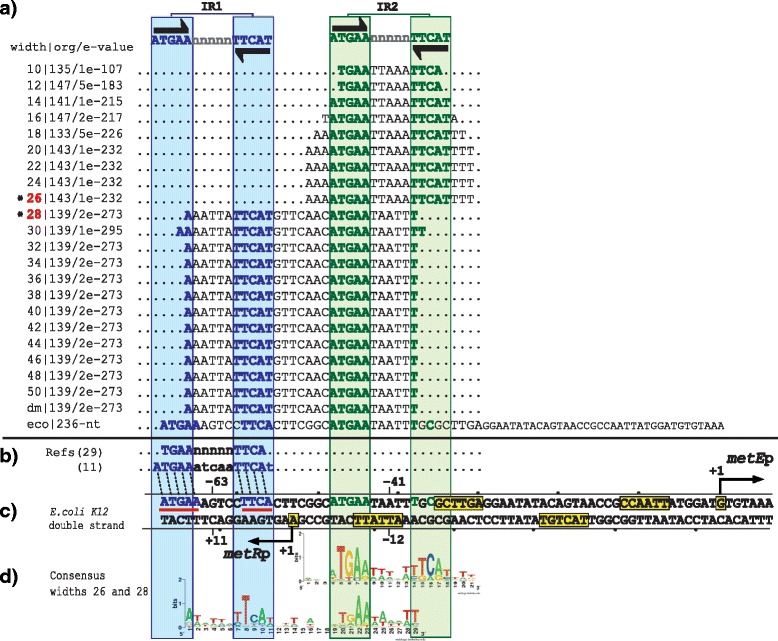
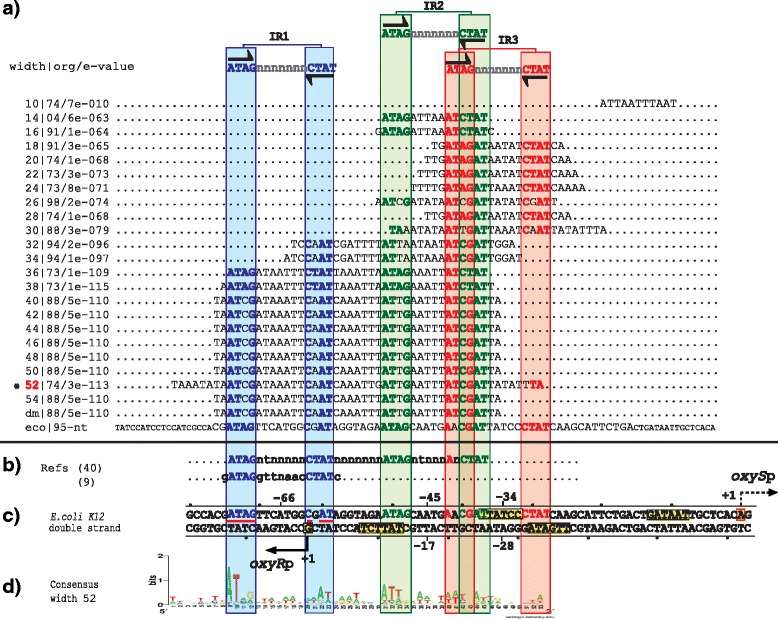
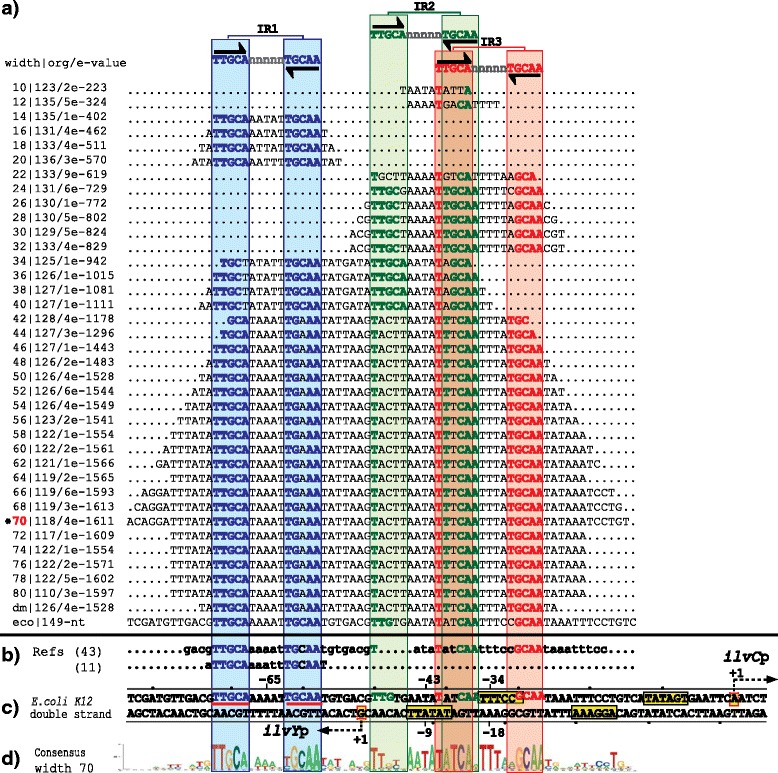
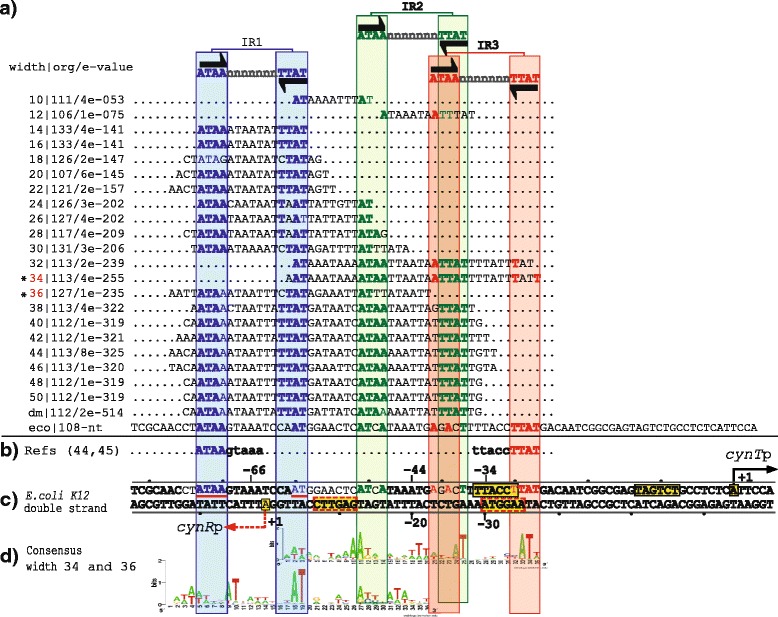
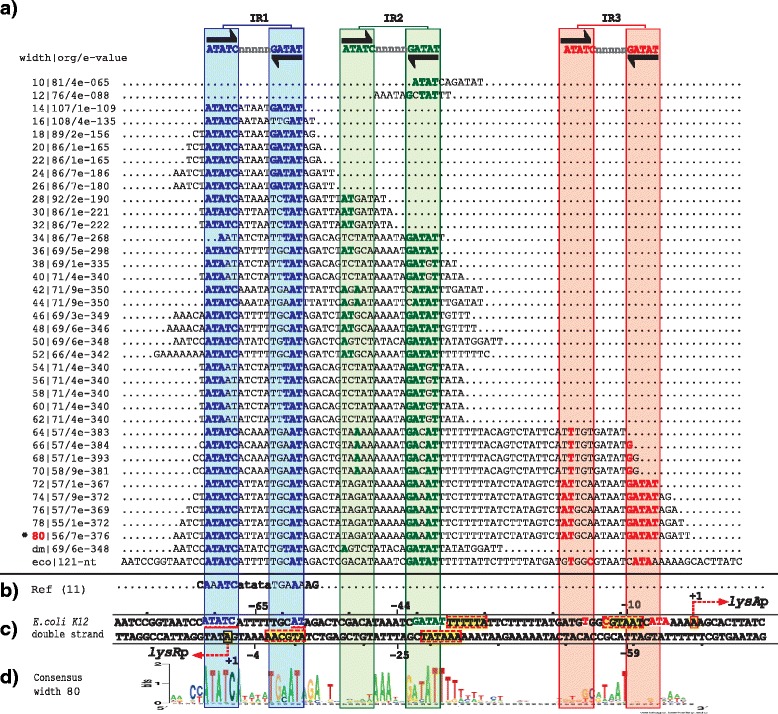
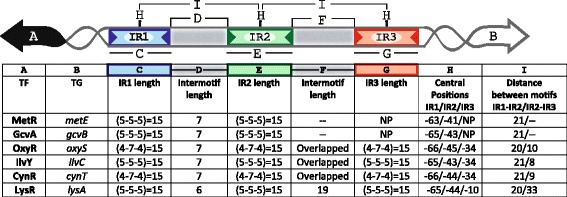
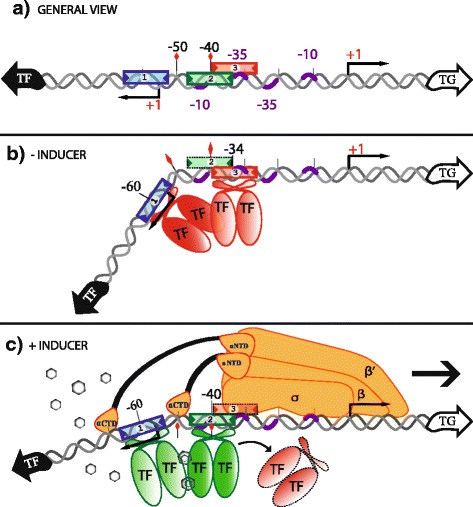
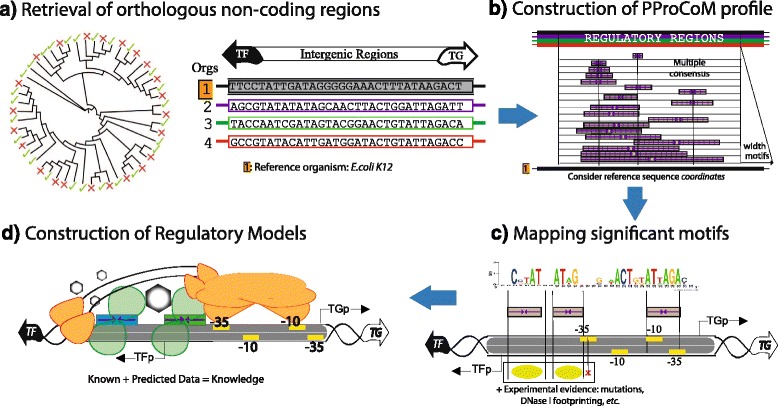
Results: We developed a computational protocol based on molecular and structural criteria to perform biologically meaningful and accurate phylogenetic footprinting analyses. Our protocol considers fundamental aspects of the TF-DNA binding process, such as: i) the active homodimeric conformations of TFs that impose symmetric structures on the TFBSs, ii) the cooperative binding of TFs, iii) the effects of the presence or absence of co-inducers, iv) the proximity between two TFBSs or one TFBS and a promoter that leads to very long spurious motifs, v) the presence of AT-rich sequences not recognized by the TF but that are required for DNA flexibility, and vi) the dynamic order in which the different binding events take place to determine a regulatory response (i.e., activation or repression). In our protocol, the abovementioned criteria were used to analyze a profile of consensus motifs generated from canonical Phylogenetic Footprinting Analyses using a set of analysis windows of incremental sizes. To evaluate the performance of our protocol, we analyzed six members of the LysR-type TF family in Gammaproteobacteria.
Conclusions: The identification of TFBSs based exclusively on the significance of the over-representation of motifs in a set of sequences might lead to inaccurate results. The consideration of different molecular and structural properties of the regulatory systems benefits the identification of TFBSs and enables the development of elaborate, biologically meaningful and precise regulatory models that offer a more integrated view of the dynamics of the regulatory process of transcription.
Keywords: Binding sites; LTTR; LysR-type transcription regulator family; Motif profile; Phylogenetic footprinting analysis; Transcription factors; Transcription regulation.
Figures

References
-
- Tagle DA, Koop BF, Goodman M, Slightom JL, Hess DL, Jones RT. Embryonic epsilon and gamma globin genes of a prosimian primate (galago crassicaudatus). Nucleotide and amino acid sequences, developmental regulation and phylogenetic footprints. J Mol Biol. 1988;203:439–55. doi: 10.1016/0022-2836(88)90011-3. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
