

Amyloid-Beta and Phosphorylated Tau Accumulations Cause Abnormalities at Synapses of Alzheimer's disease Neurons
- PMID: 27567878
- PMCID: PMC5793225
- DOI: 10.3233/JAD-160612
Amyloid-Beta and Phosphorylated Tau Accumulations Cause Abnormalities at Synapses of Alzheimer's disease Neurons
Abstract
Amyloid-beta (Aβ) and hyperphosphorylated tau are hallmark lesions of Alzheimer's disease (AD). However, the loss of synapses and dysfunctions of neurotransmission are more directly tied to disease severity. The role of these lesions in the pathoetiological progression of the disease remains contested. Biochemical, cellular, molecular, and pathological studies provided several lines of evidence and improved our understanding of how Aβ and hyperphosphorylated tau accumulation may directly harm synapses and alter neurotransmission. In vitro evidence suggests that Aβ and hyperphosphorylated tau have both direct and indirect cytotoxic effects that affect neurotransmission, axonal transport, signaling cascades, organelle function, and immune response in ways that lead to synaptic loss and dysfunctions in neurotransmitter release. Observations in preclinical models and autopsy studies support these findings, suggesting that while the pathoetiology of positive lesions remains elusive, their removal may reduce disease severity and progression. The purpose of this article is to highlight the need for further investigation of the role of tau in disease progression and its interactions with Aβ and neurotransmitters alike.
Keywords: Amyloid; neurotransmitters; synapse; tau.
Figures
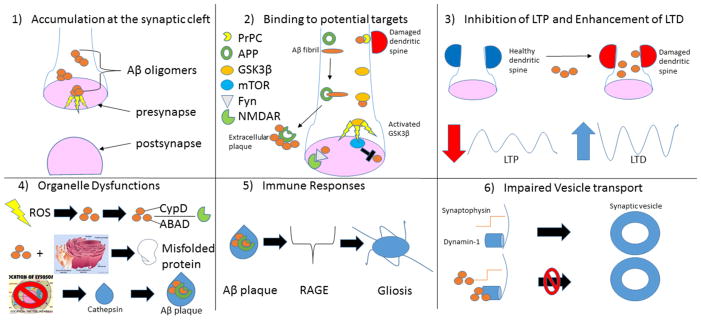
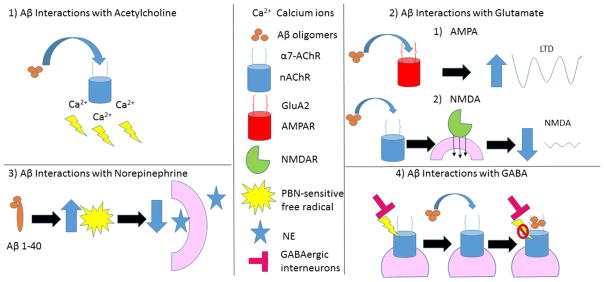
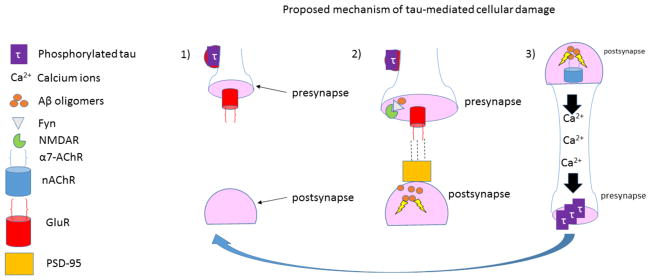
References
-
- DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer disease: correlation with cognitive severity. Annals of neurology. 1990;27(5):457–464. - PubMed
-
- Scheff SW, DeKosky ST, Price DA. Quantitative assessment of cortical synaptic density in Alzheimer disease. Neurobiology of aging. 1990;11(1):29–37. - PubMed
-
- Scheff SW, Price DA. Synapse loss in the temporal lobe in Alzheimer disease. Annals of neurology. 1993;33(2):190–199. - PubMed
-
- Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 2007;68(18):1501–1508. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
