

Impaired Presynaptic High-Affinity Choline Transporter Causes a Congenital Myasthenic Syndrome with Episodic Apnea
- PMID: 27569547
- PMCID: PMC5011057
- DOI: 10.1016/j.ajhg.2016.06.033
Impaired Presynaptic High-Affinity Choline Transporter Causes a Congenital Myasthenic Syndrome with Episodic Apnea
Abstract
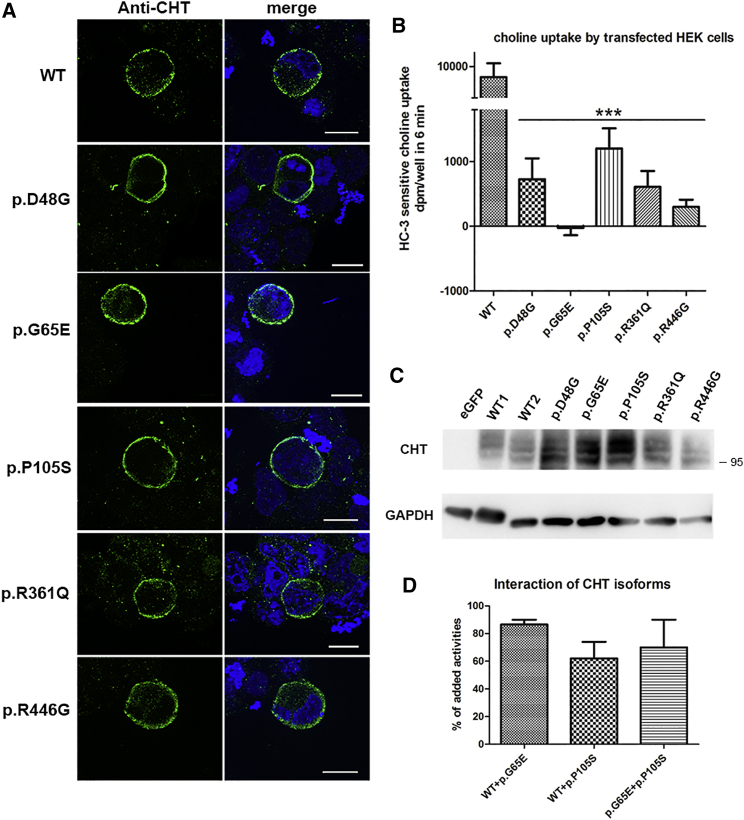
The neuromuscular junction (NMJ) is one of the best-studied cholinergic synapses. Inherited defects of peripheral neurotransmission result in congenital myasthenic syndromes (CMSs), a clinically and genetically heterogeneous group of rare diseases with fluctuating fatigable muscle weakness as the clinical hallmark. Whole-exome sequencing and Sanger sequencing in six unrelated families identified compound heterozygous and homozygous mutations in SLC5A7 encoding the presynaptic sodium-dependent high-affinity choline transporter 1 (CHT), which is known to be mutated in one dominant form of distal motor neuronopathy (DHMN7A). We identified 11 recessive mutations in SLC5A7 that were associated with a spectrum of severe muscle weakness ranging from a lethal antenatal form of arthrogryposis and severe hypotonia to a neonatal form of CMS with episodic apnea and a favorable prognosis when well managed at the clinical level. As expected given the critical role of CHT for multisystemic cholinergic neurotransmission, autonomic dysfunctions were reported in the antenatal form and cognitive impairment was noticed in half of the persons with the neonatal form. The missense mutations induced a near complete loss of function of CHT activity in cell models. At the human NMJ, a delay in synaptic maturation and an altered maintenance were observed in the antenatal and neonatal forms, respectively. Increased synaptic expression of butyrylcholinesterase was also observed, exposing the dysfunction of cholinergic metabolism when CHT is deficient in vivo. This work broadens the clinical spectrum of human diseases resulting from reduced CHT activity and highlights the complexity of cholinergic metabolism at the synapse.
Copyright © 2016 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures


References
-
- Black S.A., Rylett R.J. Choline transporter CHT regulation and function in cholinergic neurons. Cent. Nerv. Syst. Agents Med. Chem. 2012;12:114–121. - PubMed
-
- Haga T. Molecular properties of the high-affinity choline transporter CHT1. J. Biochem. 2014;156:181–194. - PubMed
-
- Byring R.F., Pihko H., Tsujino A., Shen X.M., Gustafsson B., Hackman P., Ohno K., Engel A.G., Udd B. Congenital myasthenic syndrome associated with episodic apnea and sudden infant death. Neuromuscul. Disord. 2002;12:548–553. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
