

RNA Splicing Modulation Selectively Impairs Leukemia Stem Cell Maintenance in Secondary Human AML
- PMID: 27570067
- PMCID: PMC5097015
- DOI: 10.1016/j.stem.2016.08.003
RNA Splicing Modulation Selectively Impairs Leukemia Stem Cell Maintenance in Secondary Human AML
Abstract
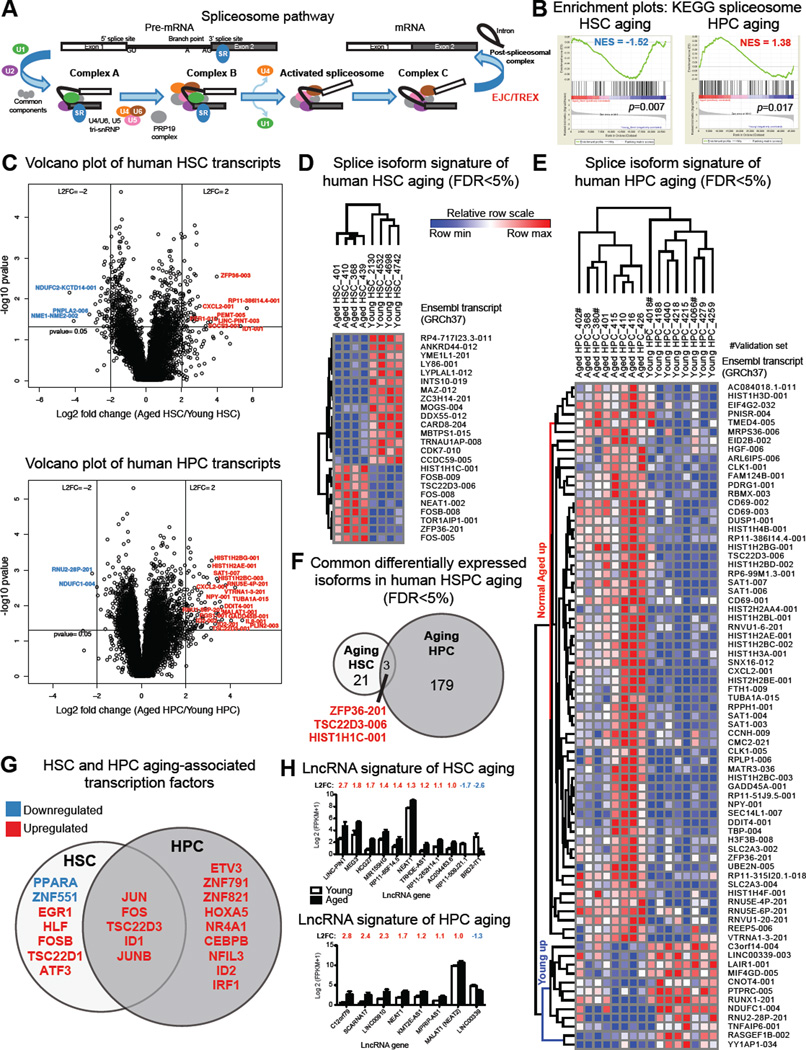
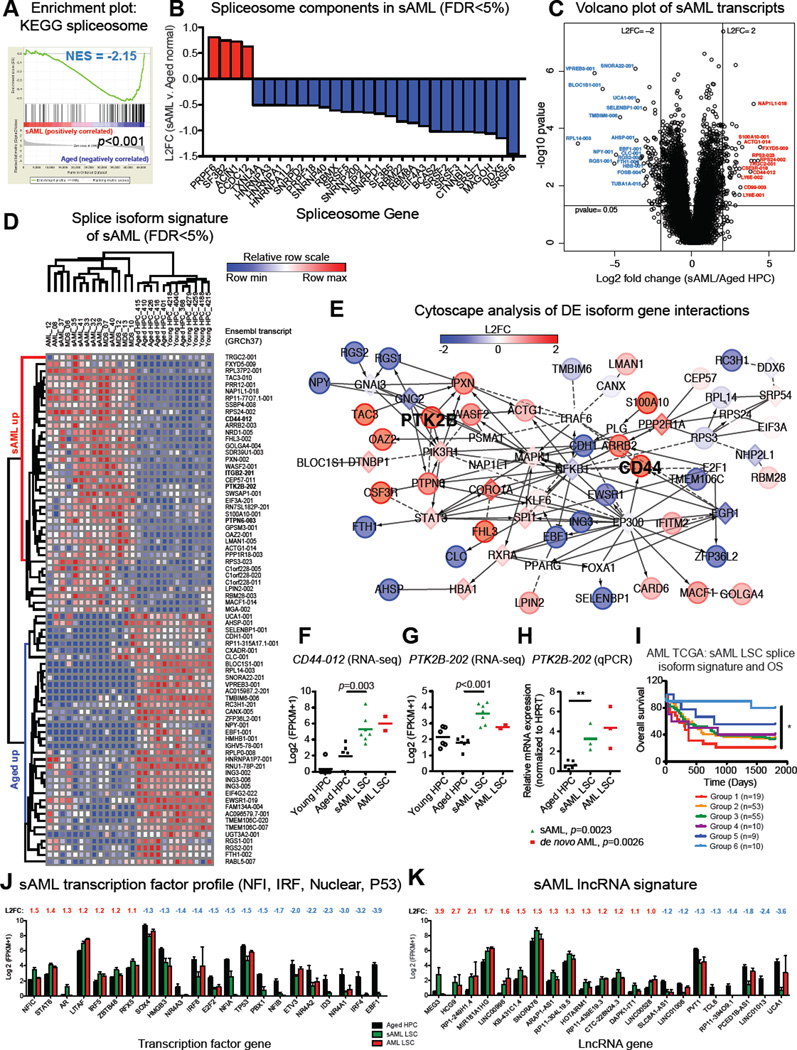
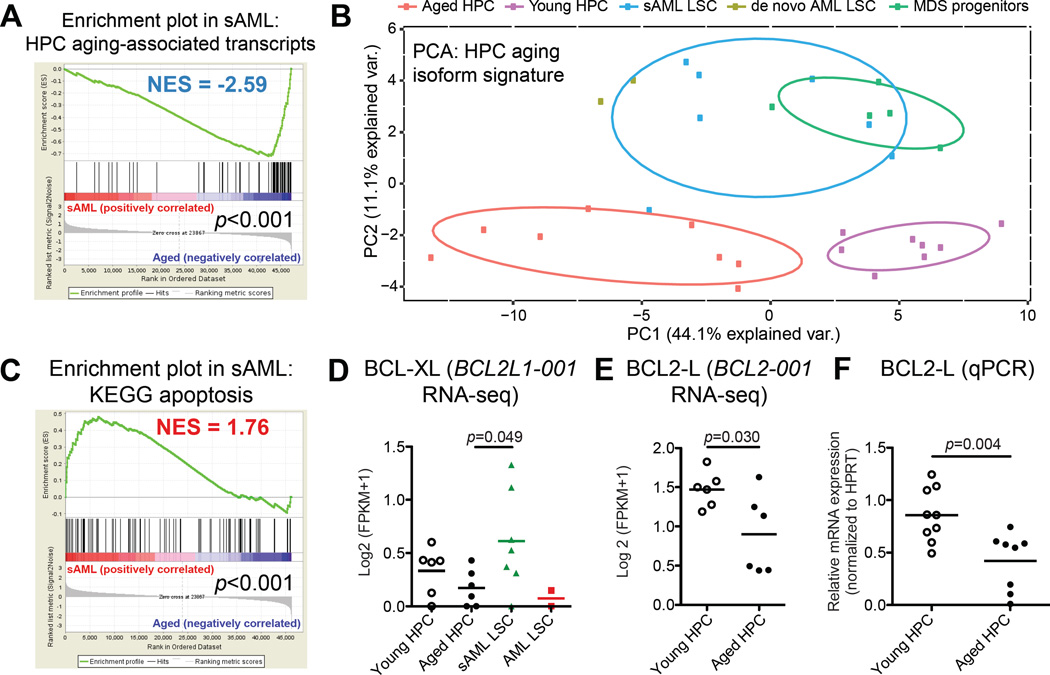
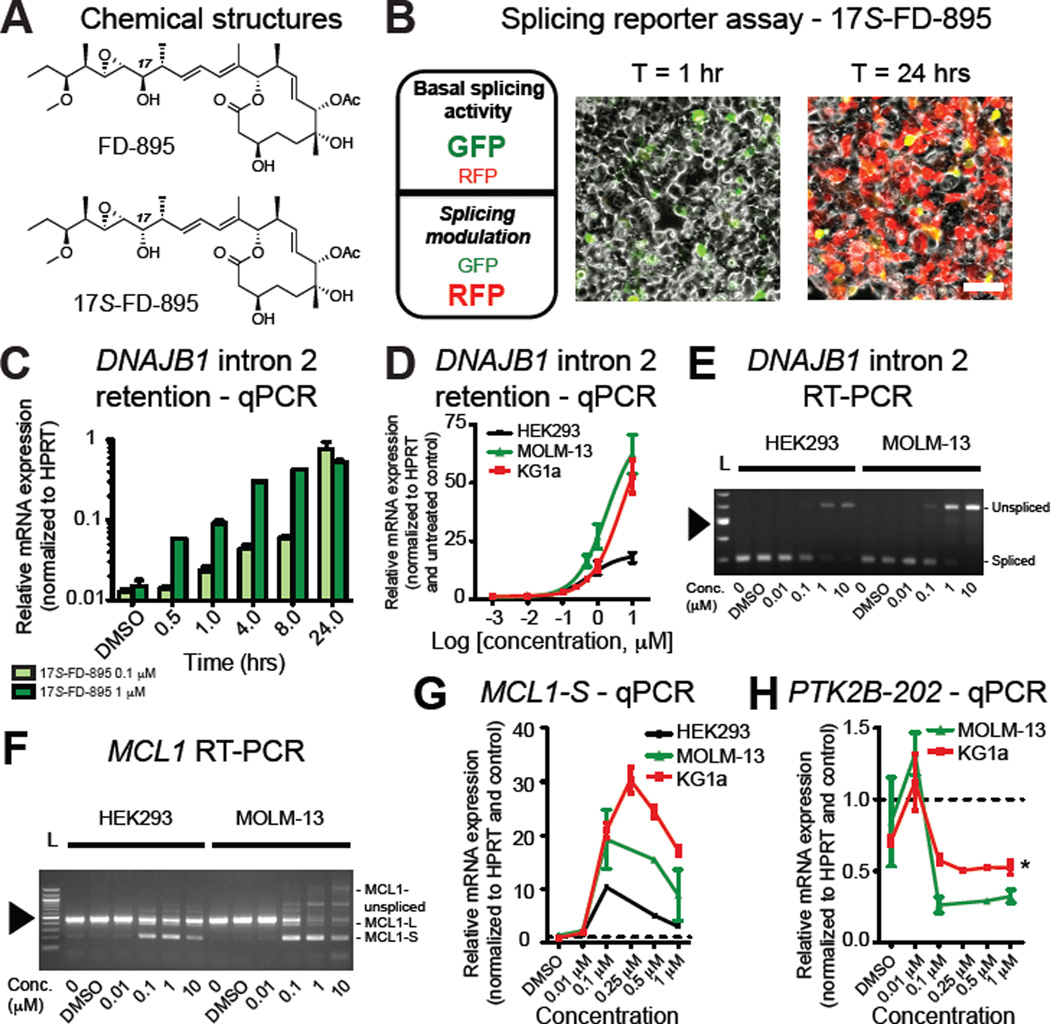
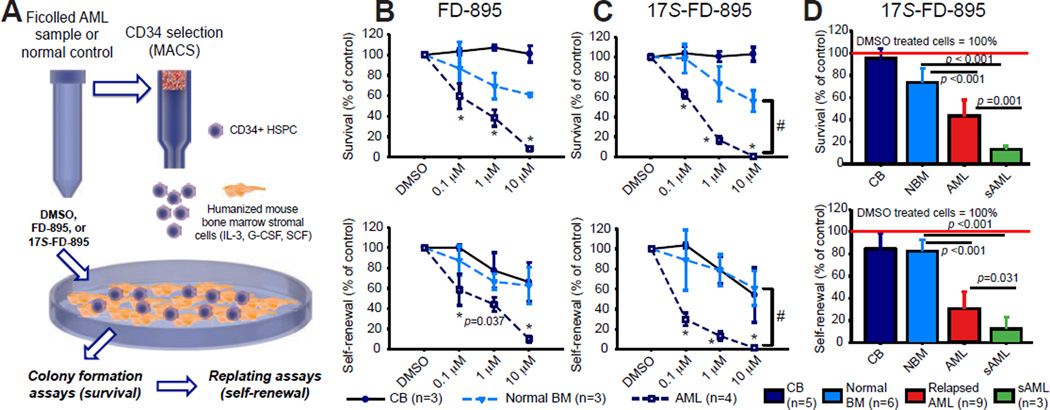
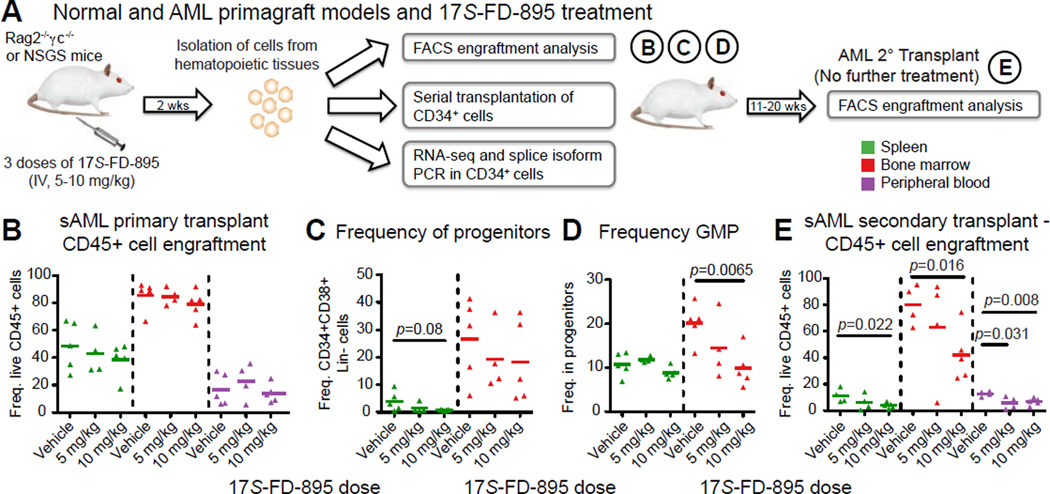
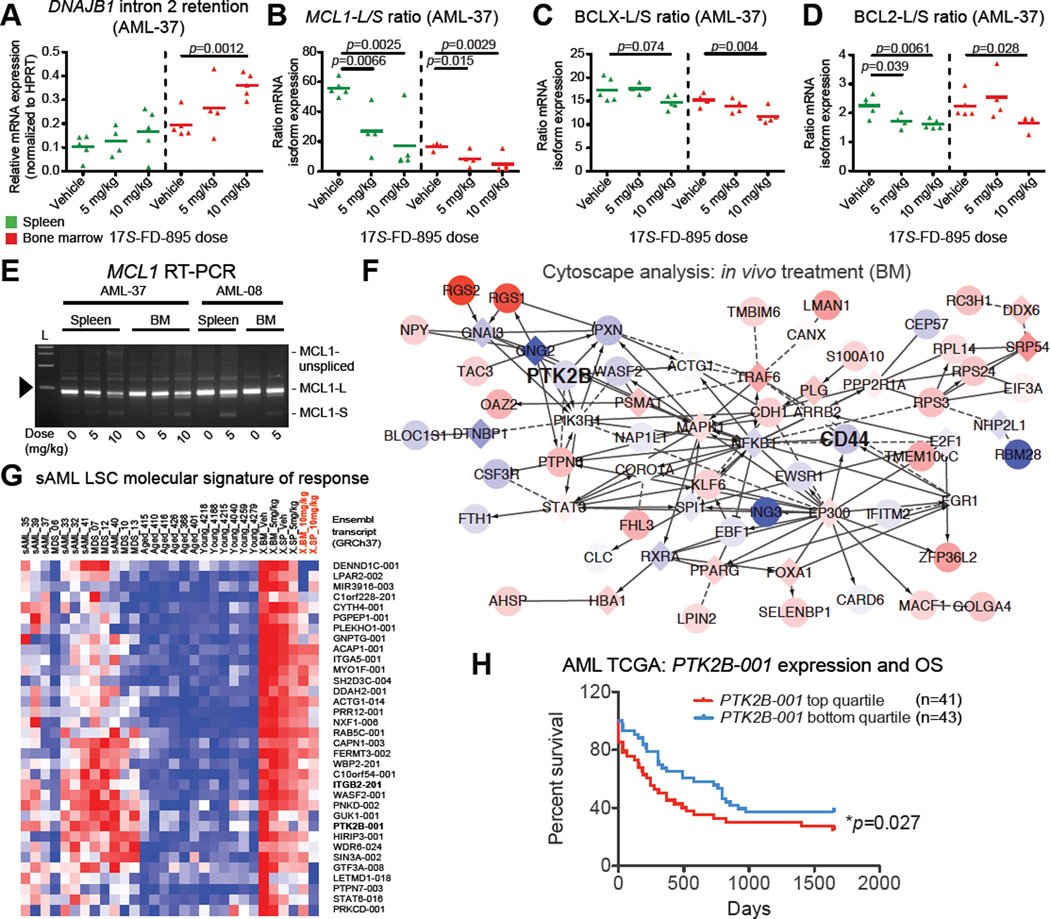
Age-related human hematopoietic stem cell (HSC) exhaustion and myeloid-lineage skewing promote oncogenic transformation of hematopoietic progenitor cells into therapy-resistant leukemia stem cells (LSCs) in secondary acute myeloid leukemia (AML). While acquisition of clonal DNA mutations has been linked to increased rates of secondary AML for individuals older than 60 years, the contribution of RNA processing alterations to human hematopoietic stem and progenitor aging and LSC generation remains unclear. Comprehensive RNA sequencing and splice-isoform-specific PCR uncovered characteristic RNA splice isoform expression patterns that distinguished normal young and aged human stem and progenitor cells (HSPCs) from malignant myelodysplastic syndrome (MDS) and AML progenitors. In splicing reporter assays and pre-clinical patient-derived AML models, treatment with a pharmacologic splicing modulator, 17S-FD-895, reversed pro-survival splice isoform switching and significantly impaired LSC maintenance. Therapeutic splicing modulation, together with monitoring splice isoform biomarkers of healthy HSPC aging versus LSC generation, may be employed safely and effectively to prevent relapse, the leading cause of leukemia-related mortality.
Copyright © 2016 Elsevier Inc. All rights reserved.
Conflict of interest statement
Authors have no competing financial interests to disclose.
Figures
Comment in
-
Leukemic Stem Cells S(p)liced Off.Cell Stem Cell. 2016 Nov 3;19(5):561-563. doi: 10.1016/j.stem.2016.10.016. Cell Stem Cell. 2016. PMID: 27814474
-
Aberrant RNA splicing and mutations in spliceosome complex in acute myeloid leukemia.Stem Cell Investig. 2017 Feb 9;4:6. doi: 10.21037/sci.2017.01.06. eCollection 2017. Stem Cell Investig. 2017. PMID: 28217708 Free PMC article.
References
-
- Adamia S, Haibe-Kains B, Pilarski PM, Bar-Natan M, Pevzner S, Avet-Loiseau H, Lode L, Verselis S, Fox EA, Burke J, et al. A genome-wide aberrant RNA splicing in patients with acute myeloid leukemia identifies novel potential disease markers and therapeutic targets. Clin Cancer Res. 2014;20:1135–1145. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
