

The metabolic background is a global player in Saccharomyces gene expression epistasis
- PMID: 27572163
- PMCID: PMC5131842
- DOI: 10.1038/nmicrobiol.2015.30
The metabolic background is a global player in Saccharomyces gene expression epistasis
Abstract
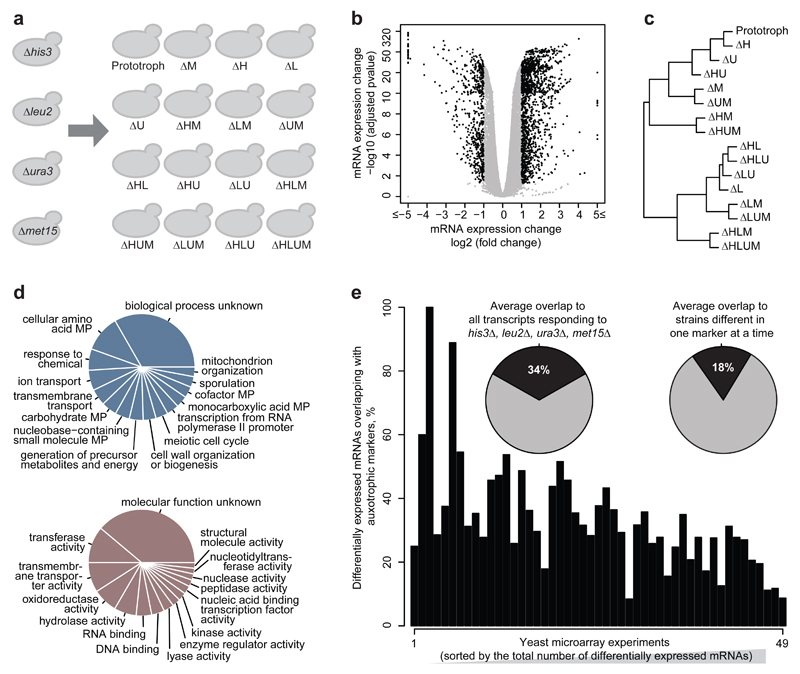
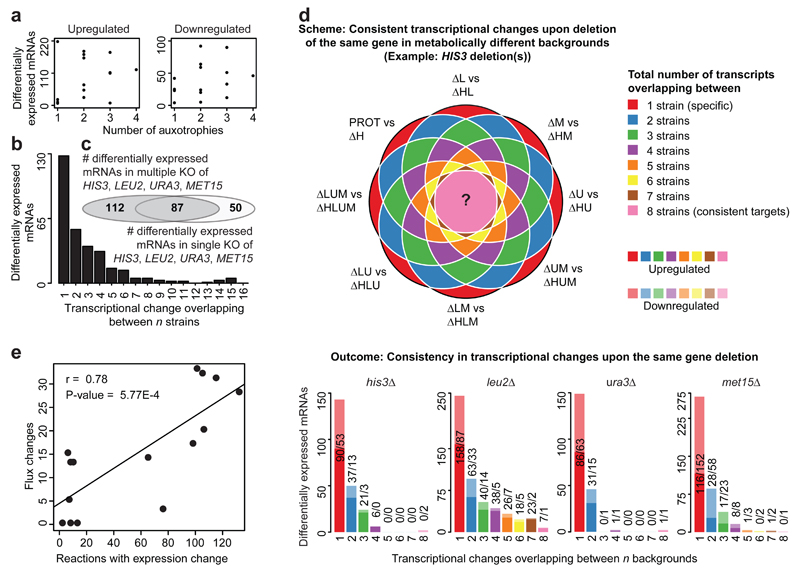
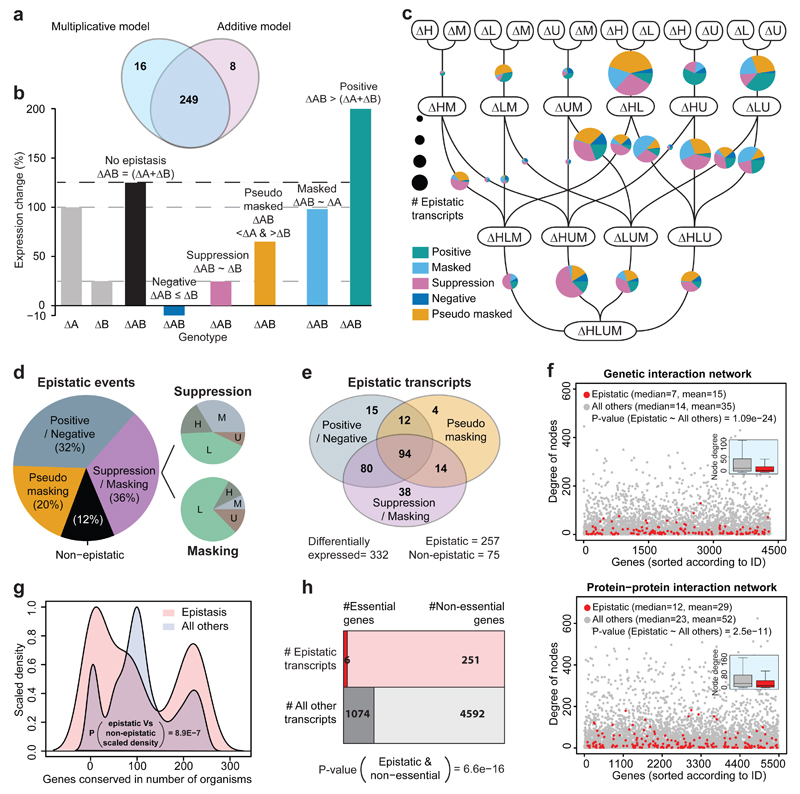
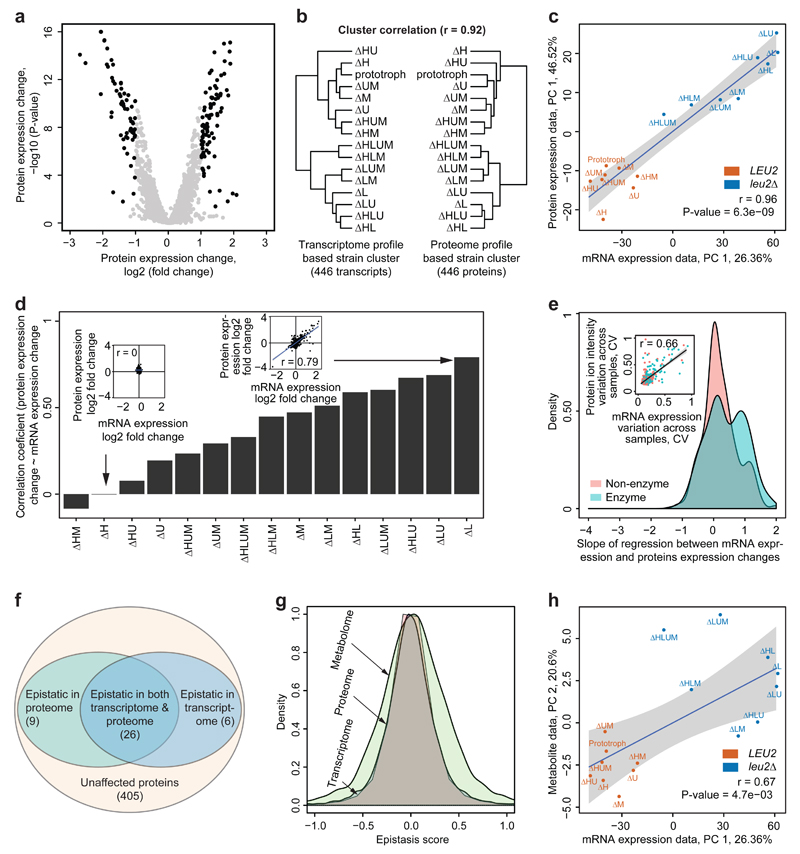
The regulation of gene expression in response to nutrient availability is fundamental to the genotype-phenotype relationship. The metabolic-genetic make-up of the cell, as reflected in auxotrophy, is hence likely to be a determinant of gene expression. Here, we address the importance of the metabolic-genetic background by monitoring transcriptome, proteome and metabolome in a repertoire of 16 Saccharomyces cerevisiae laboratory backgrounds, combinatorially perturbed in histidine, leucine, methionine and uracil biosynthesis. The metabolic background affected up to 85% of the coding genome. Suggesting widespread confounding, these transcriptional changes show, on average, 83% overlap between unrelated auxotrophs and 35% with previously published transcriptomes generated for non-metabolic gene knockouts. Background-dependent gene expression correlated with metabolic flux and acted, predominantly through masking or suppression, on 88% of transcriptional interactions epistatically. As a consequence, the deletion of the same metabolic gene in a different background could provoke an entirely different transcriptional response. Propagating to the proteome and scaling up at the metabolome, metabolic background dependencies reveal the prevalence of metabolism-dependent epistasis at all regulatory levels. Urging a fundamental change of the prevailing laboratory practice of using auxotrophs and nutrient supplemented media, these results reveal epistatic intertwining of metabolism with gene expression on the genomic scale.
Conflict of interest statement
The authors declare no competitive interests.
Figures
Comment in
-
Systems biology: Supplementation is not sufficient.Nat Microbiol. 2016 Feb 24;1:16016. doi: 10.1038/nmicrobiol.2016.16. Nat Microbiol. 2016. PMID: 27572175 No abstract available.
References
-
- Albert R. Scale-free networks in cell biology. J Cell Sci. 2005;118:4947–4957. - PubMed
-
- Barabási A-L, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nat Rev Genet. 2004;5:101–113. - PubMed
-
- Jeong H, Tombor B, Albert R, Oltvai ZN, Barabási A-L. The large-scale organization of metabolic networks. Nature. 2000;407:651–654. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
