

Identifying lineage effects when controlling for population structure improves power in bacterial association studies
- PMID: 27572646
- PMCID: PMC5049680
- DOI: 10.1038/nmicrobiol.2016.41
Identifying lineage effects when controlling for population structure improves power in bacterial association studies
Abstract
Bacteria pose unique challenges for genome-wide association studies because of strong structuring into distinct strains and substantial linkage disequilibrium across the genome(1,2). Although methods developed for human studies can correct for strain structure(3,4), this risks considerable loss-of-power because genetic differences between strains often contribute substantial phenotypic variability(5). Here, we propose a new method that captures lineage-level associations even when locus-specific associations cannot be fine-mapped. We demonstrate its ability to detect genes and genetic variants underlying resistance to 17 antimicrobials in 3,144 isolates from four taxonomically diverse clonal and recombining bacteria: Mycobacterium tuberculosis, Staphylococcus aureus, Escherichia coli and Klebsiella pneumoniae. Strong selection, recombination and penetrance confer high power to recover known antimicrobial resistance mechanisms and reveal a candidate association between the outer membrane porin nmpC and cefazolin resistance in E. coli. Hence, our method pinpoints locus-specific effects where possible and boosts power by detecting lineage-level differences when fine-mapping is intractable.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
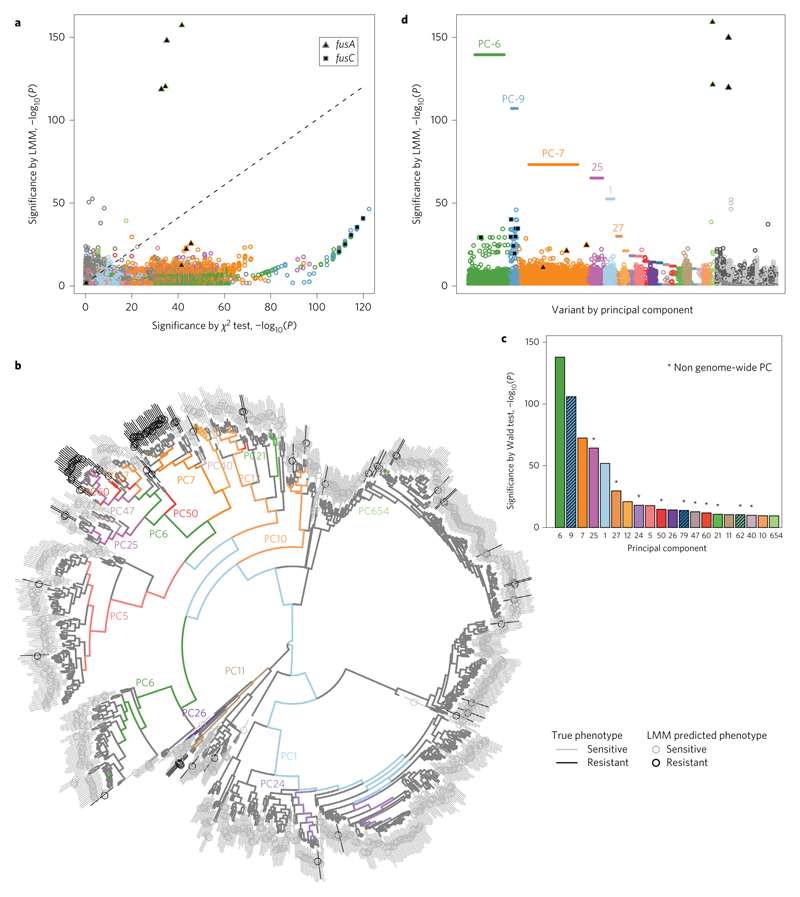
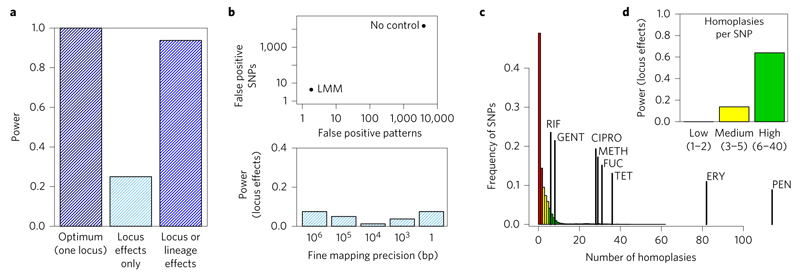
Comment in
-
Bacterial genomics: Microbial GWAS coming of age.Nat Microbiol. 2016 Apr 26;1:16059. doi: 10.1038/nmicrobiol.2016.59. Nat Microbiol. 2016. PMID: 27572652 No abstract available.
References
-
- Feil EJ, Spratt BG. Recombination and the structures of bacterial pathogens. Annu Rev Microbiol. 2001;55:561–590. - PubMed
-
- Falush D, Bowden R. Genome-wide association mapping in bacteria? Trends Microbiol. 2006;14:353–355. - PubMed
-
- Stephens M, Balding DJ. Bayesian statistical methods for genetic association studies. Nature Rev Genet. 2009;10:681–690. - PubMed
-
- Cordero OX, Polz MF. Explaining microbial genomic diversity in light of evolutionary ecology. Nature Rev Microbiol. 2014;12:263–273. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
