

Cross-species identification of genomic drivers of squamous cell carcinoma development across preneoplastic intermediates
- PMID: 27574101
- PMCID: PMC5013636
- DOI: 10.1038/ncomms12601
Cross-species identification of genomic drivers of squamous cell carcinoma development across preneoplastic intermediates
Abstract
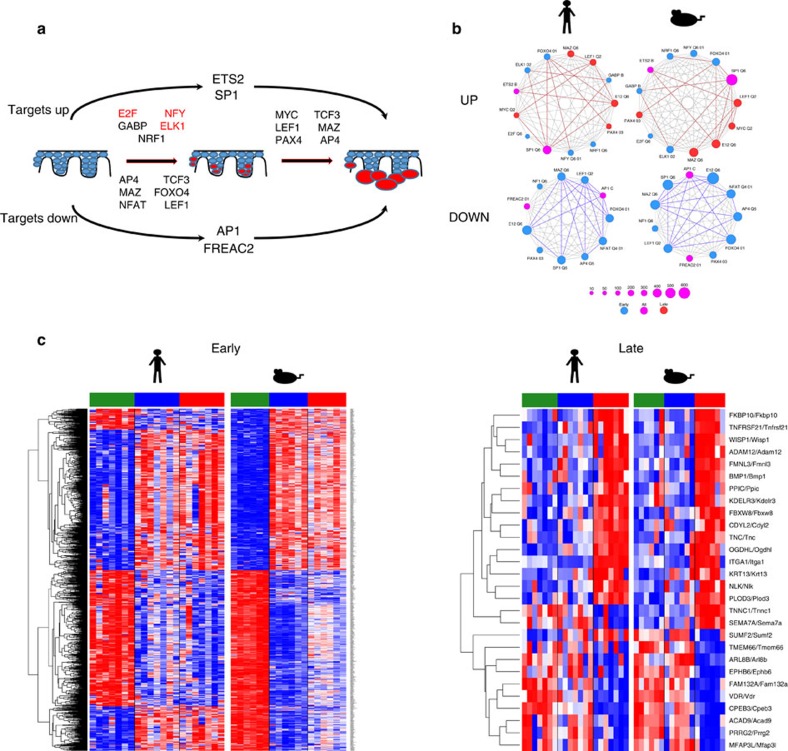
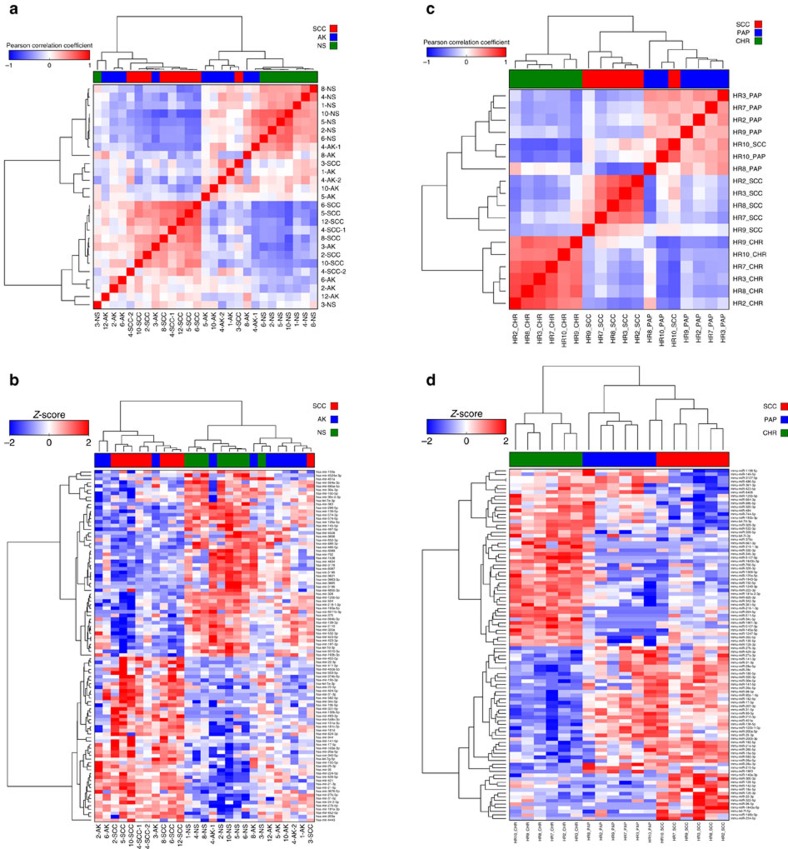
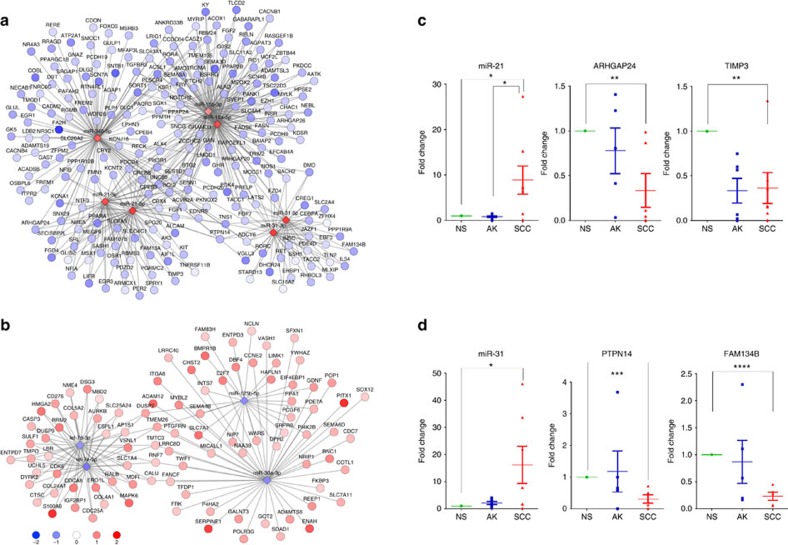
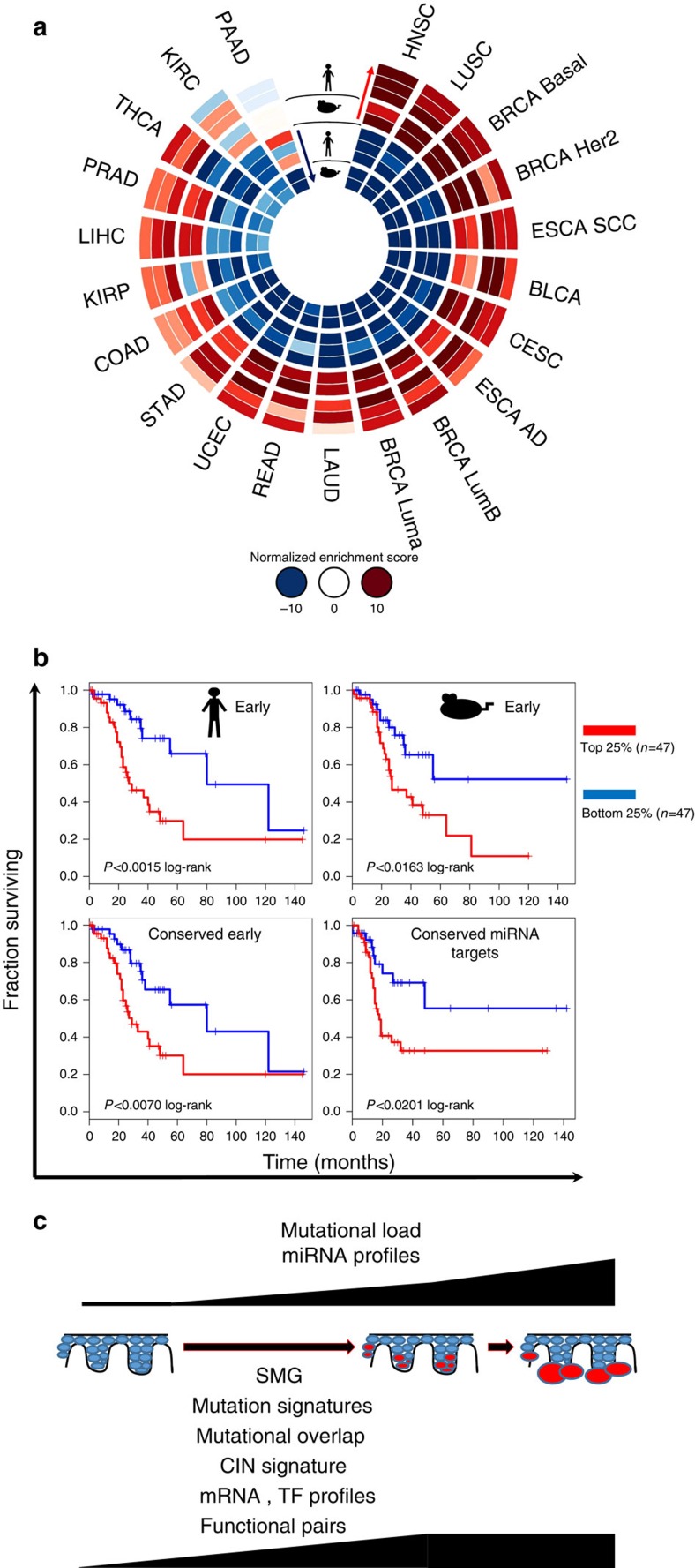
Cutaneous squamous cell carcinoma (cuSCC) comprises 15-20% of all skin cancers, accounting for over 700,000 cases in USA annually. Most cuSCC arise in association with a distinct precancerous lesion, the actinic keratosis (AK). To identify potential targets for molecularly targeted chemoprevention, here we perform integrated cross-species genomic analysis of cuSCC development through the preneoplastic AK stage using matched human samples and a solar ultraviolet radiation-driven Hairless mouse model. We identify the major transcriptional drivers of this progression sequence, showing that the key genomic changes in cuSCC development occur in the normal skin to AK transition. Our data validate the use of this ultraviolet radiation-driven mouse cuSCC model for cross-species analysis and demonstrate that cuSCC bears deep molecular similarities to multiple carcinogen-driven SCCs from diverse sites, suggesting that cuSCC may serve as an effective, accessible model for multiple SCC types and that common treatment and prevention strategies may be feasible.
Figures
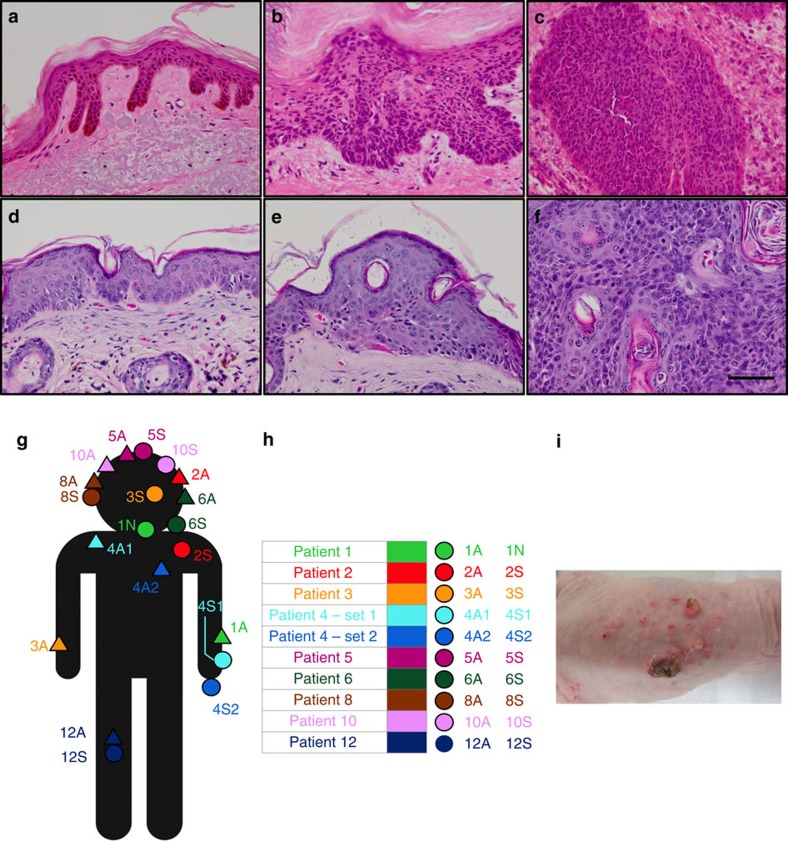
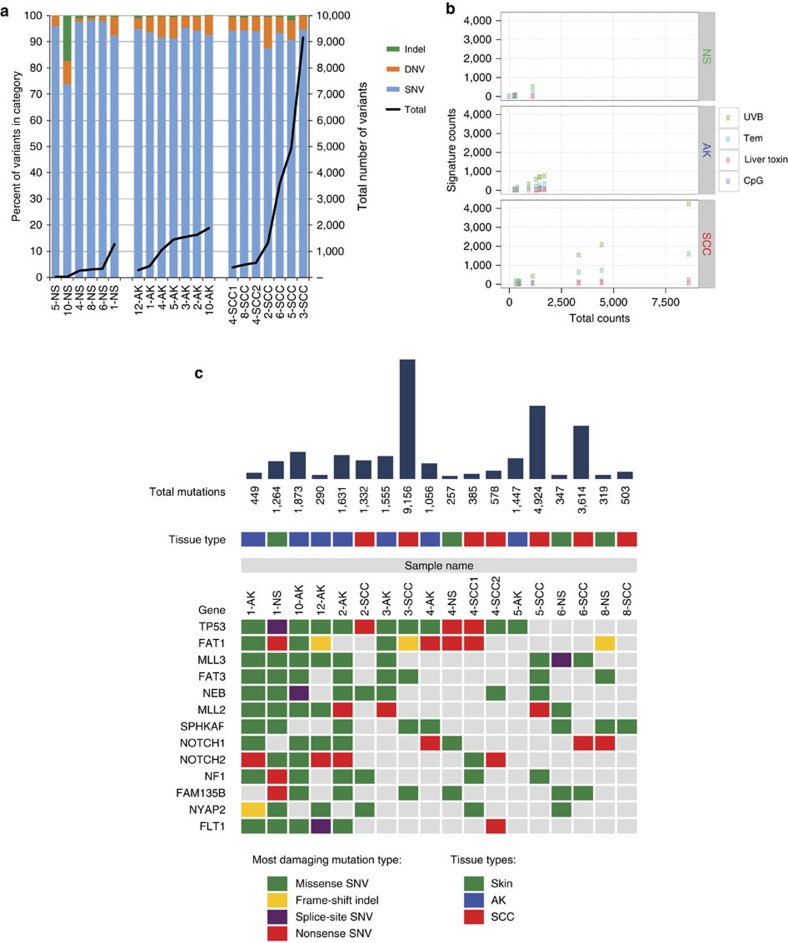
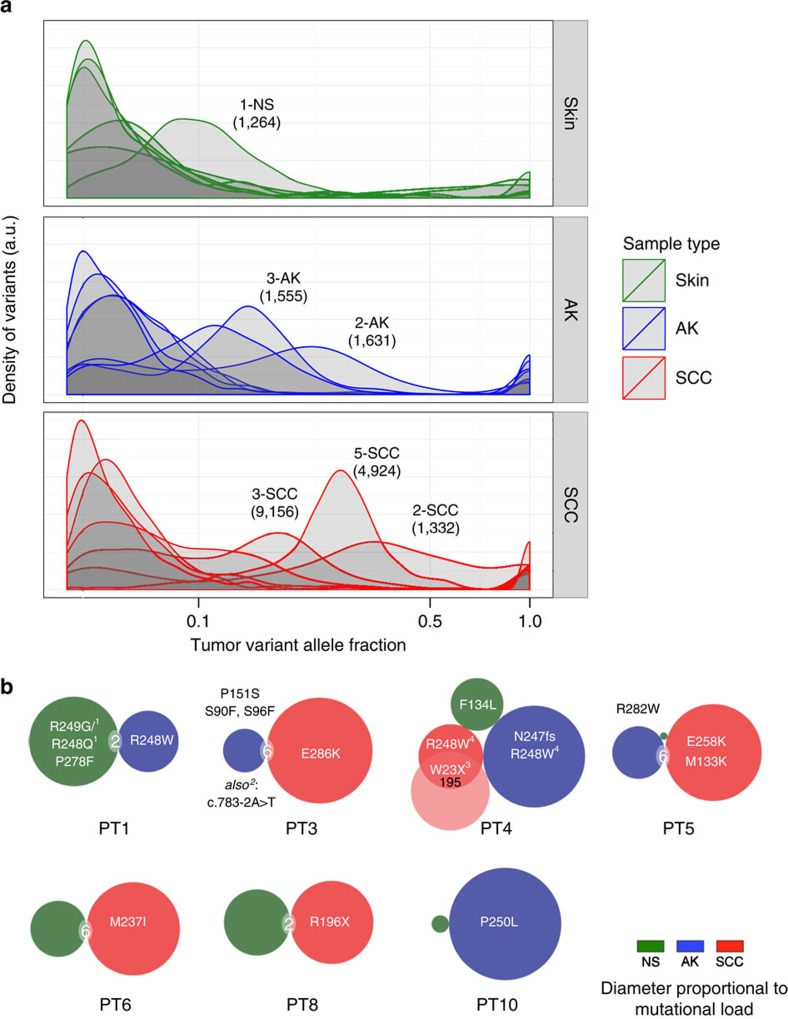
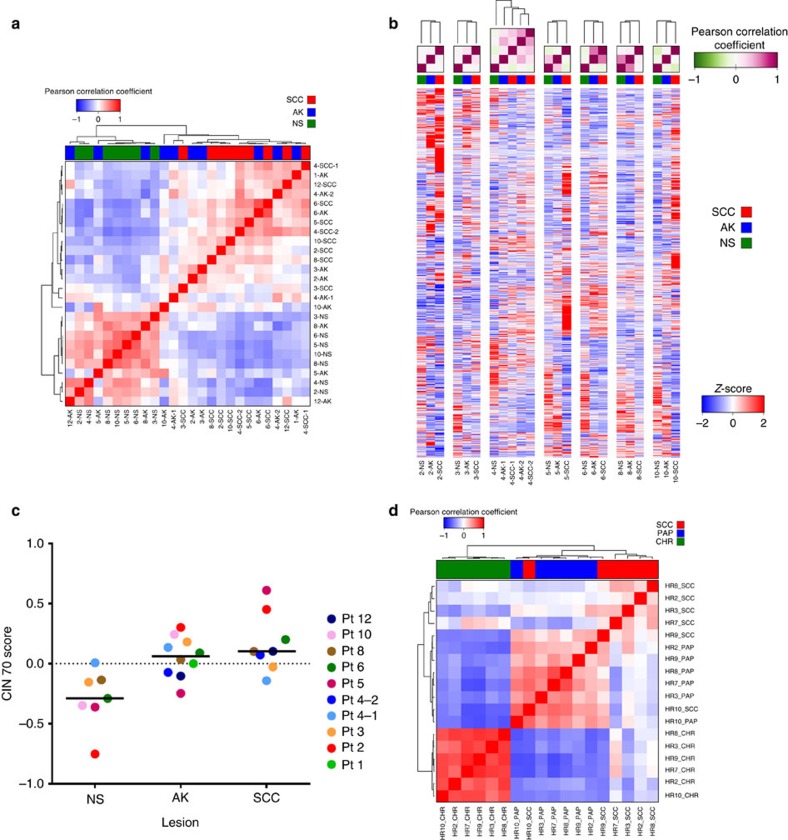
References
-
- Rosen T. & Lebwohl M. G. Prevalence and awareness of actinic keratosis: barriers and opportunities. J. Am. Acad. Dermatol. 68, S2–S9 (2013). - PubMed
-
- Warino L. et al.. Frequency and cost of actinic keratosis treatment. Dermatol. Surg. 32, 1045–1049 (2006). - PubMed
-
- Criscione V. D. et al.. Actinic keratoses: natural history and risk of malignant transformation in the Veterans Affairs Topical Tretinoin Chemoprevention Trial. Cancer 115, 2523–2530 (2009). - PubMed
-
- Karia P. S., Han J. & Schmults C. D. Cutaneous squamous cell carcinoma: estimated incidence of disease, nodal metastasis, and deaths from disease in the United States, 2012. J. Am. Acad. Dermatol. 68, 957–966 (2013). - PubMed
-
- Neidecker M. V., Davis-Ajami M. L., Balkrishnan R. & Feldman S. R. Pharmacoeconomic considerations in treating actinic keratosis. Pharmacoeconomics 27, 451–464 (2009). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
