

Efficacy of ATR inhibitors as single agents in Ewing sarcoma
- PMID: 27577084
- PMCID: PMC5312273
- DOI: 10.18632/oncotarget.11643
Efficacy of ATR inhibitors as single agents in Ewing sarcoma
Abstract
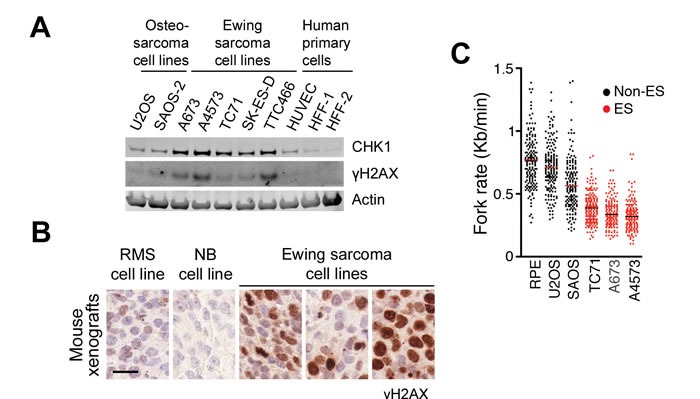
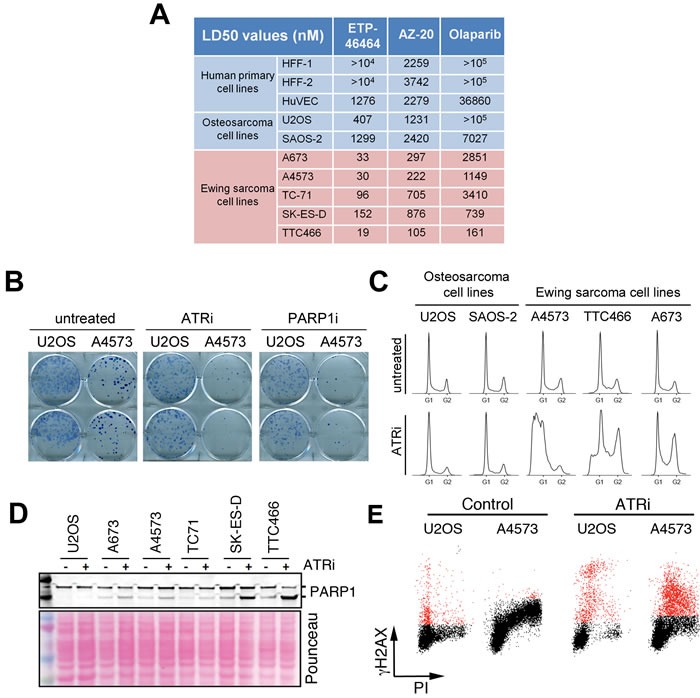
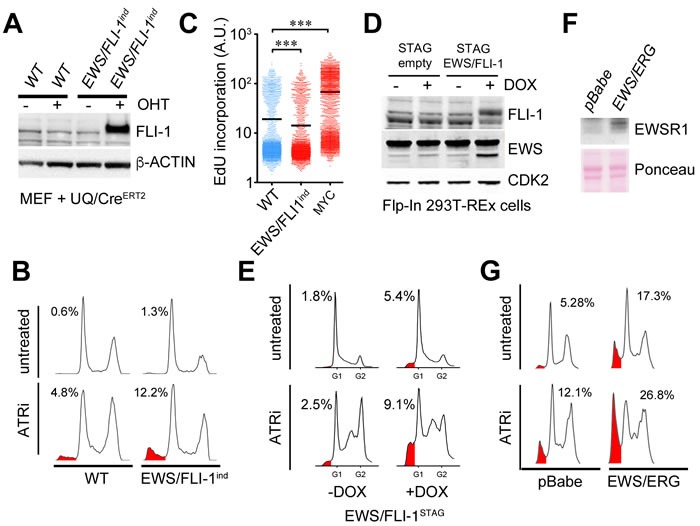
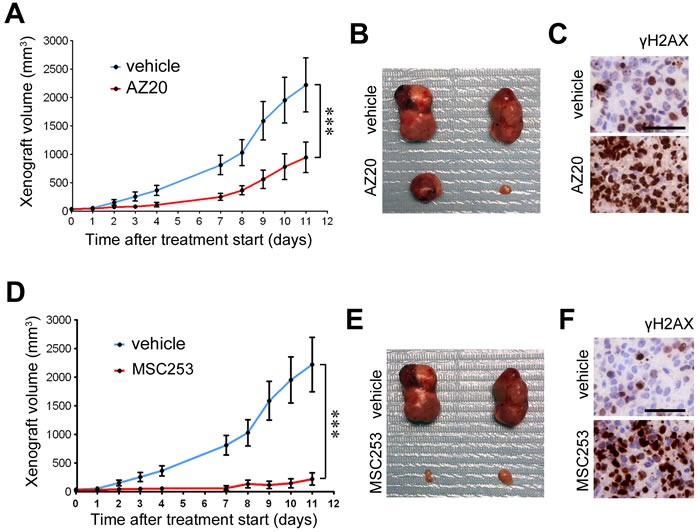
Ewing sarcomas (ES) are pediatric bone tumors that arise from a driver translocation, most frequently EWS/FLI1. Current ES treatment involves DNA damaging agents, yet the basis for the sensitivity to these therapies remains unknown. Oncogene-induced replication stress (RS) is a known source of endogenous DNA damage in cancer, which is suppressed by ATR and CHK1 kinases. We here show that ES suffer from high endogenous levels of RS, rendering them particularly dependent on the ATR pathway. Accordingly, two independent ATR inhibitors show in vitro toxicity in ES cell lines as well as in vivo efficacy in ES xenografts as single agents. Expression of EWS/FLI1 or EWS/ERG oncogenic translocations sensitizes non-ES cells to ATR inhibitors. Our data shed light onto the sensitivity of ES to genotoxic agents, and identify ATR inhibitors as a potential therapy for Ewing Sarcomas.
Keywords: ATR; DNA repair; Ewing sarcoma; cancer; replication stress.
Conflict of interest statement
O.F. receives consulting fees from Merck KGaA. The rest of the authors have no competing financial interests to disclose.
Figures
References
-
- Boveri T. Zur Frage der Entstehung Maligner Tumoren. Gustav Fischer, Jena. 1914:1–64.
-
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. - PubMed
-
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. - PubMed
-
- Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319(5868):1352–1355. - PubMed
-
- Murga M, Campaner S, Lopez-Contreras AJ, Toledo LI, Soria R, Montana MF, D'Artista L, Schleker T, Guerra C, Garcia E, Barbacid M, Hidalgo M, Amati B, Fernandez-Capetillo O. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat Struct Mol Biol. 2011;18(12):1331–1335. - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
