

Single-cell analysis of mixed-lineage states leading to a binary cell fate choice
- PMID: 27580035
- PMCID: PMC5161694
- DOI: 10.1038/nature19348
Single-cell analysis of mixed-lineage states leading to a binary cell fate choice
Erratum in
-
Author Correction: Single-cell analysis of mixed-lineage states leading to a binary cell fate choice.Nature. 2019 May;569(7755):E3. doi: 10.1038/s41586-019-1107-5. Nature. 2019. PMID: 31019298 Free PMC article.
Abstract
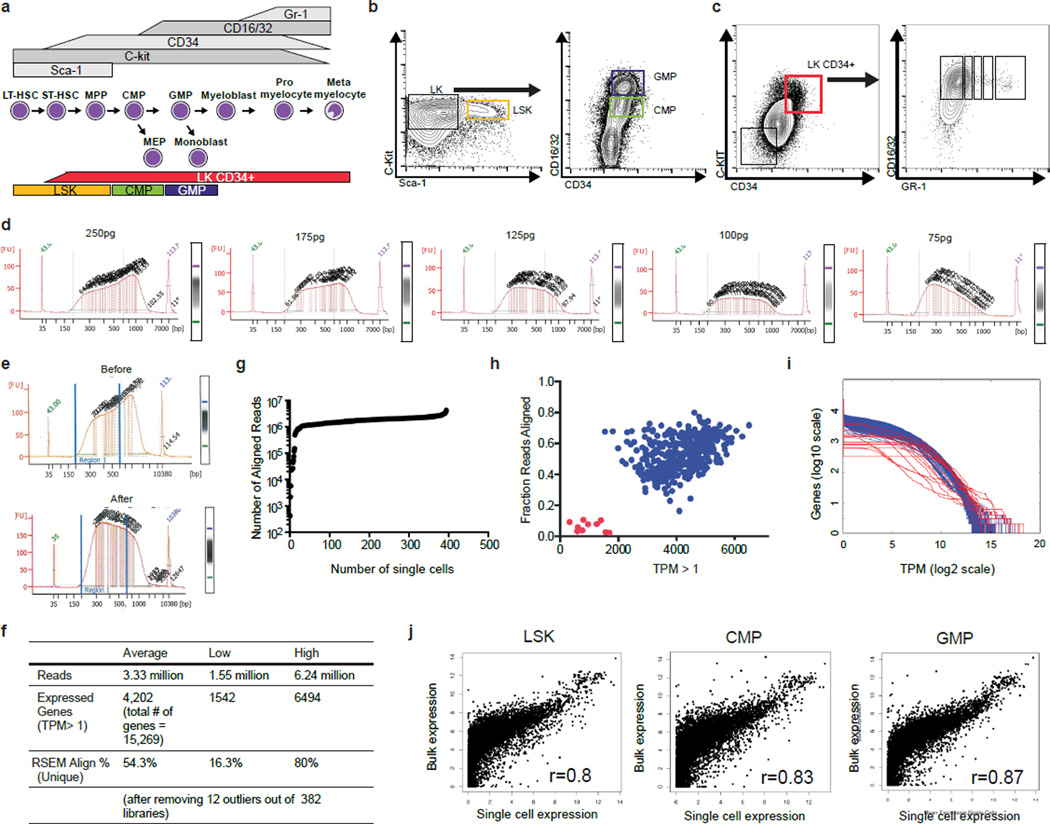
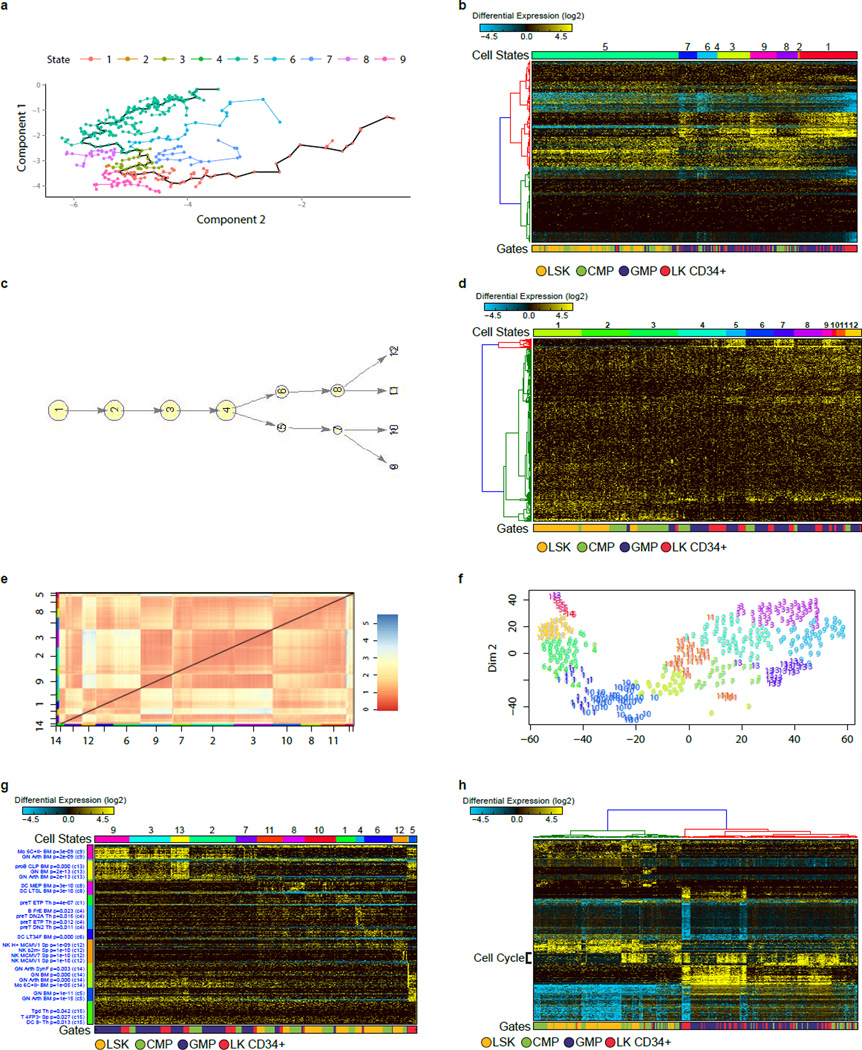
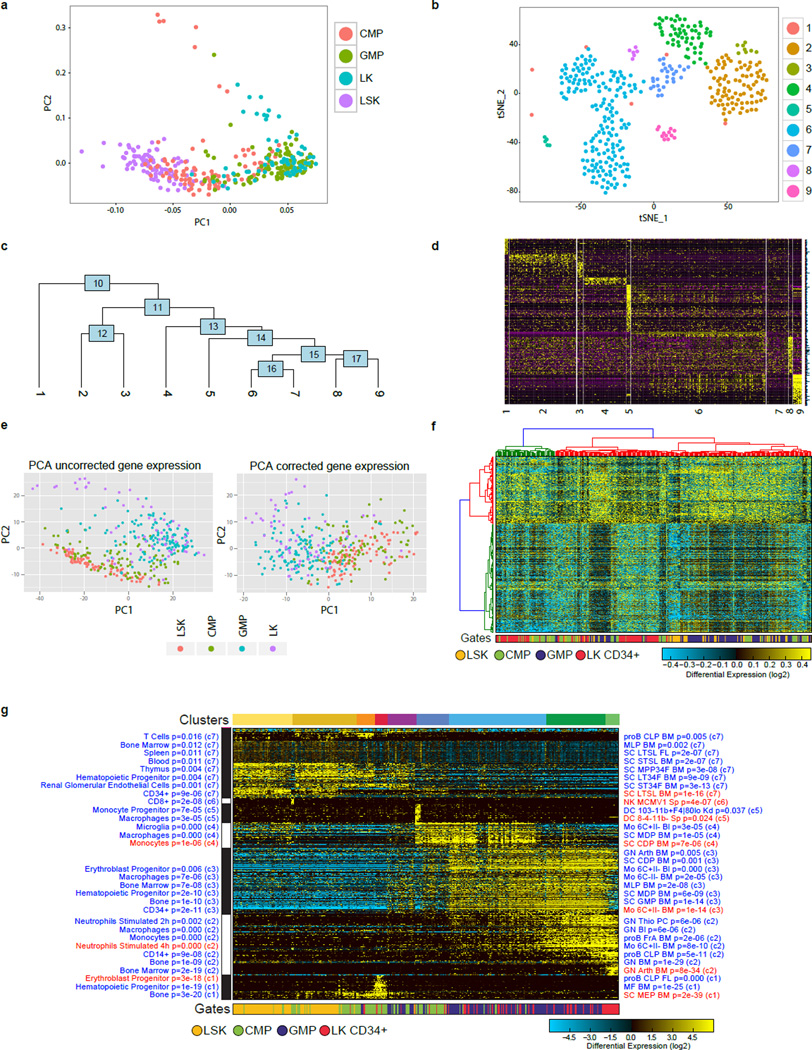
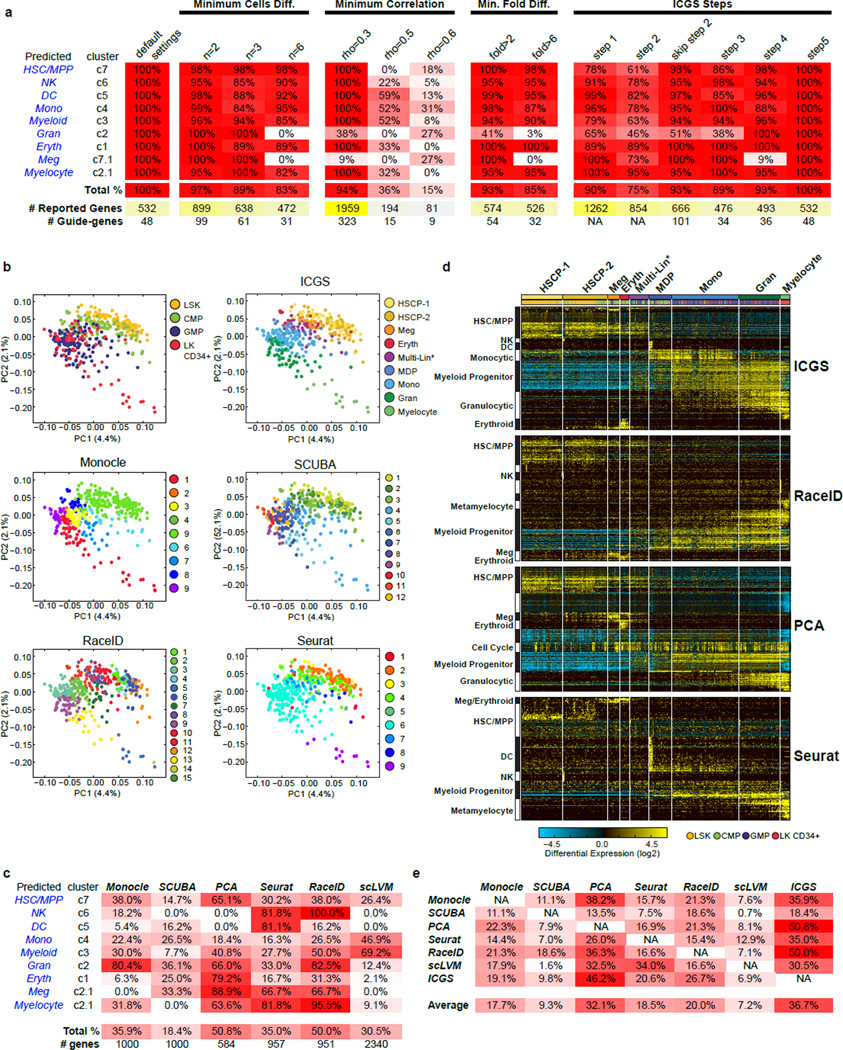
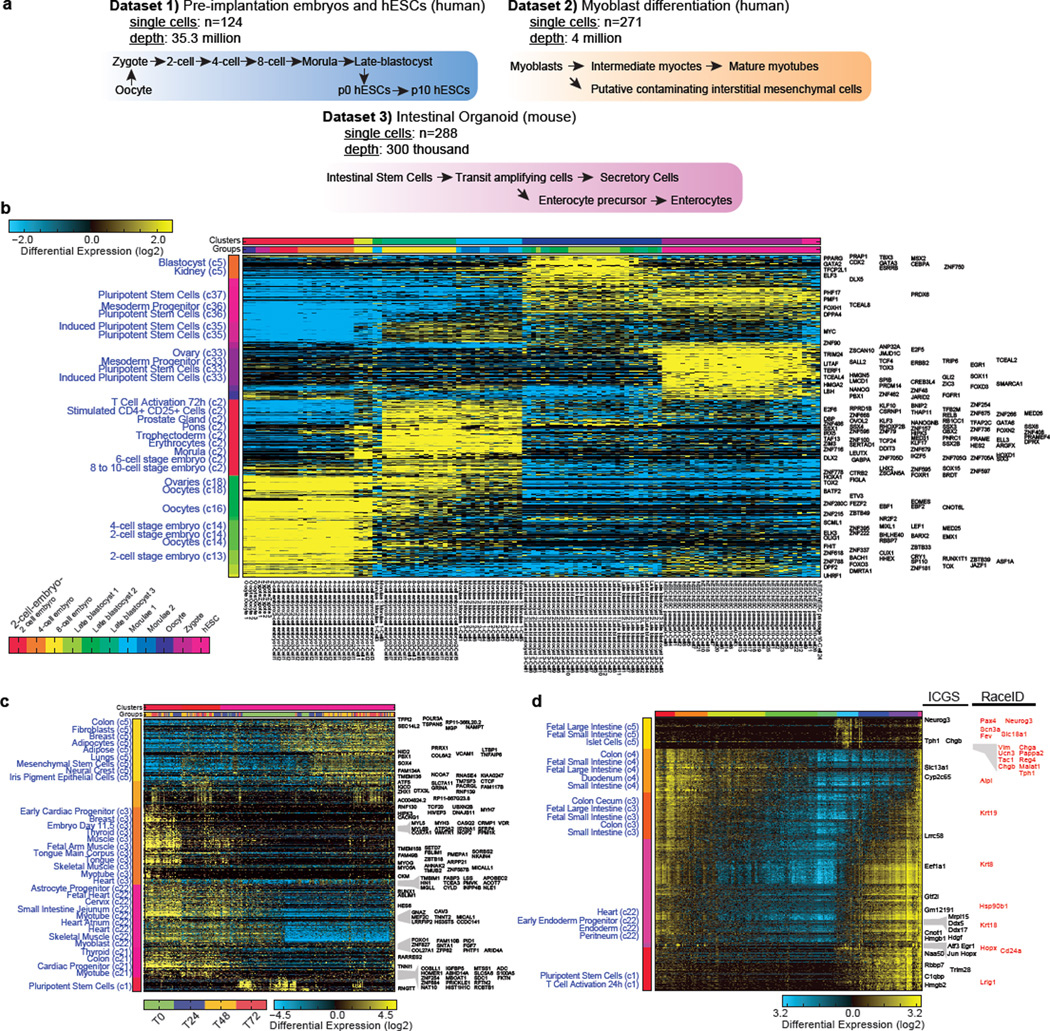
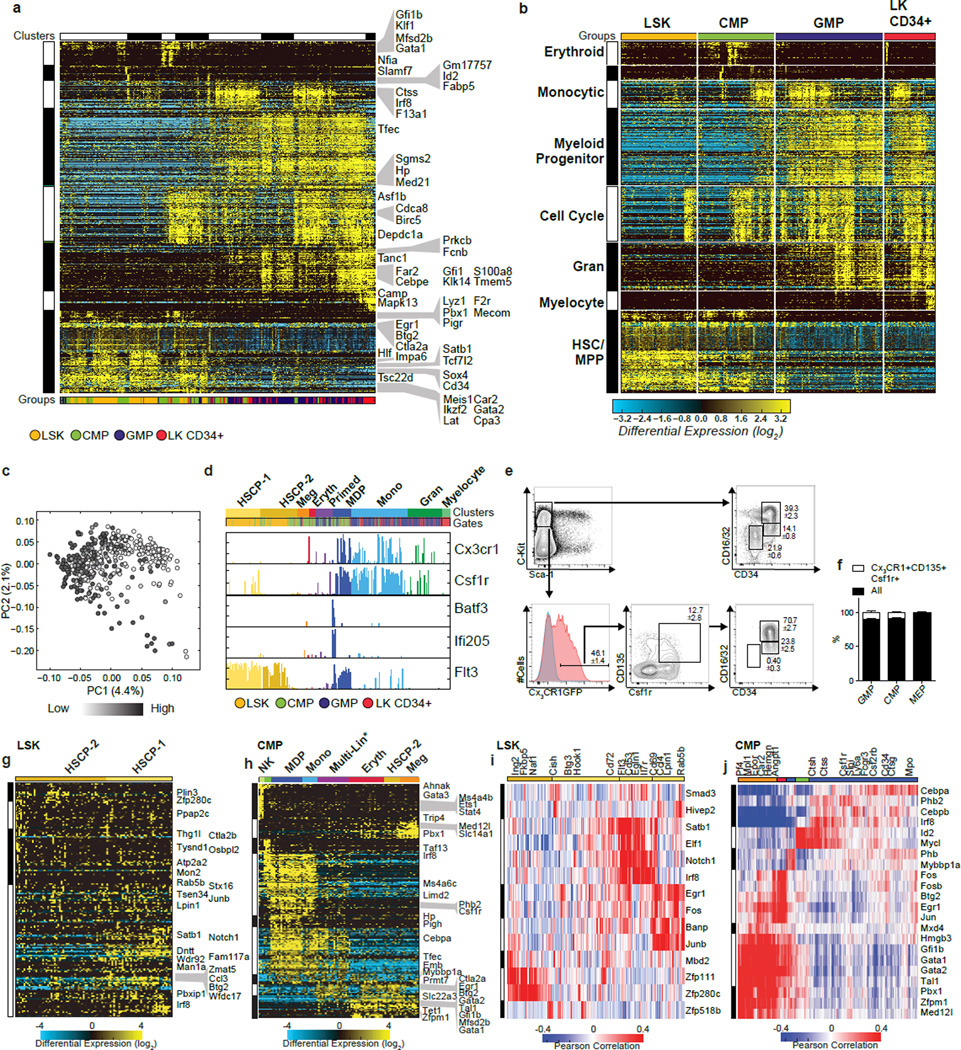
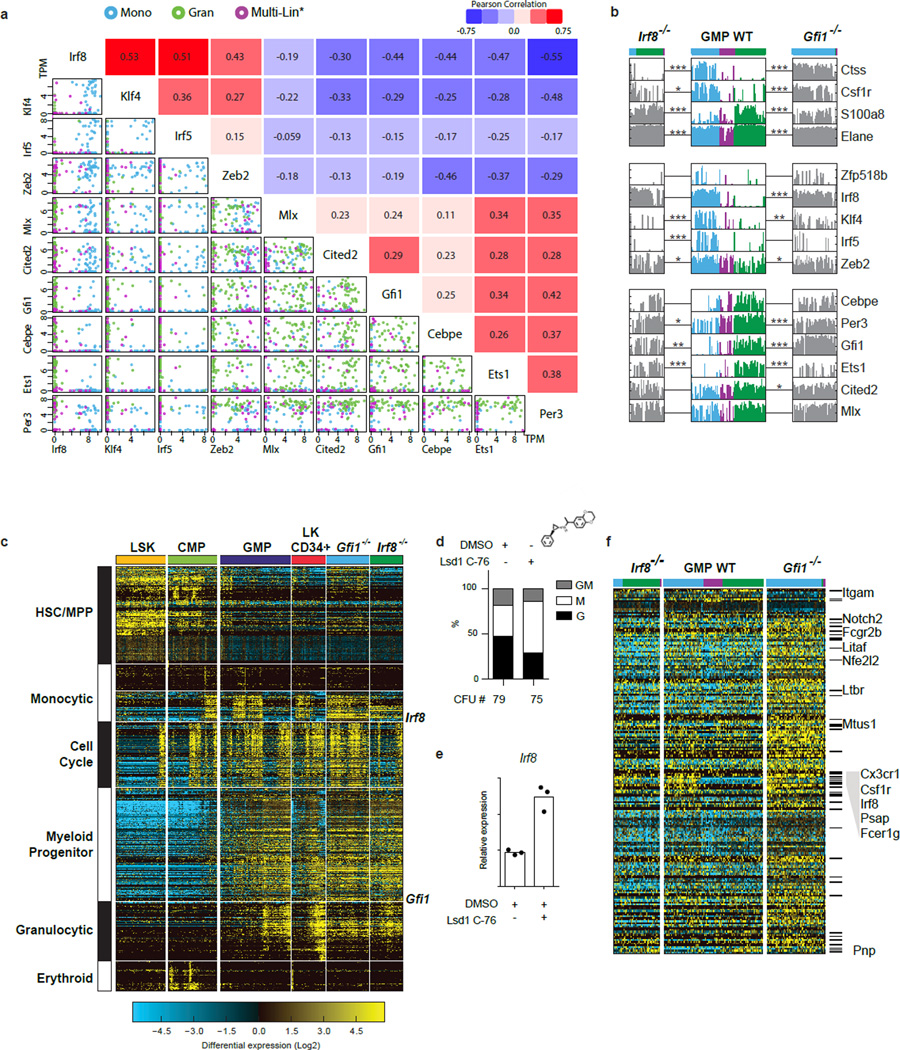
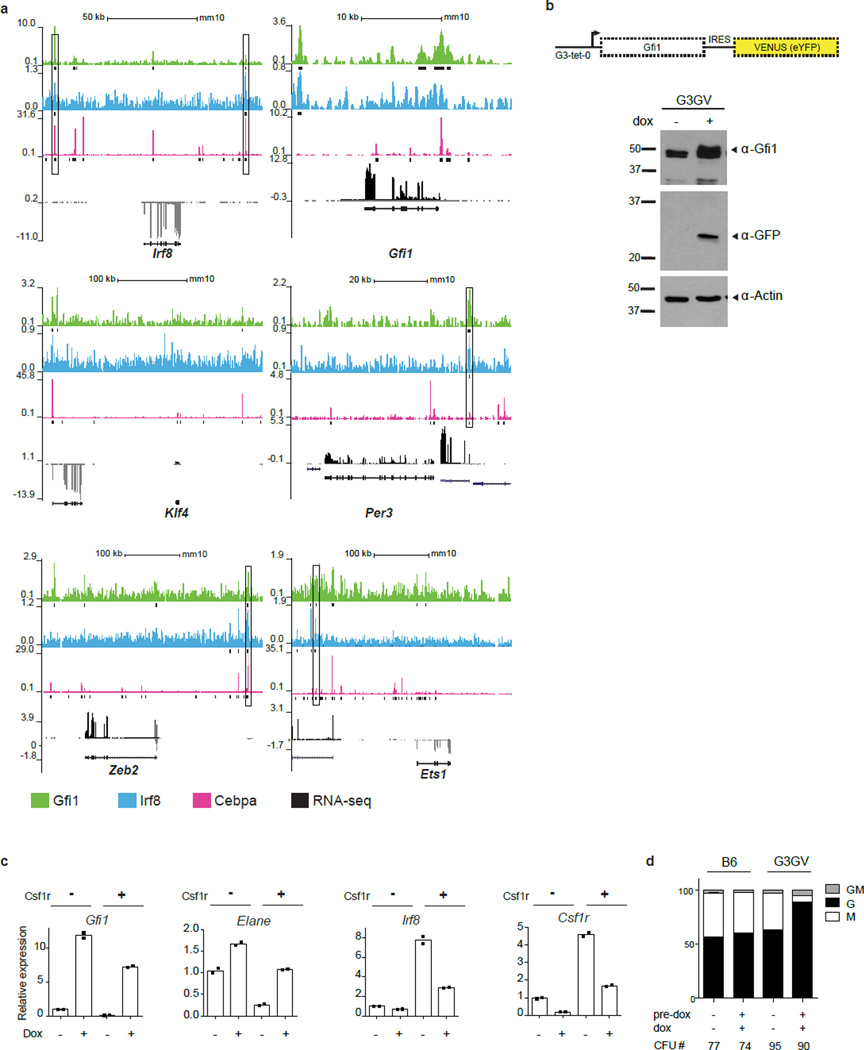
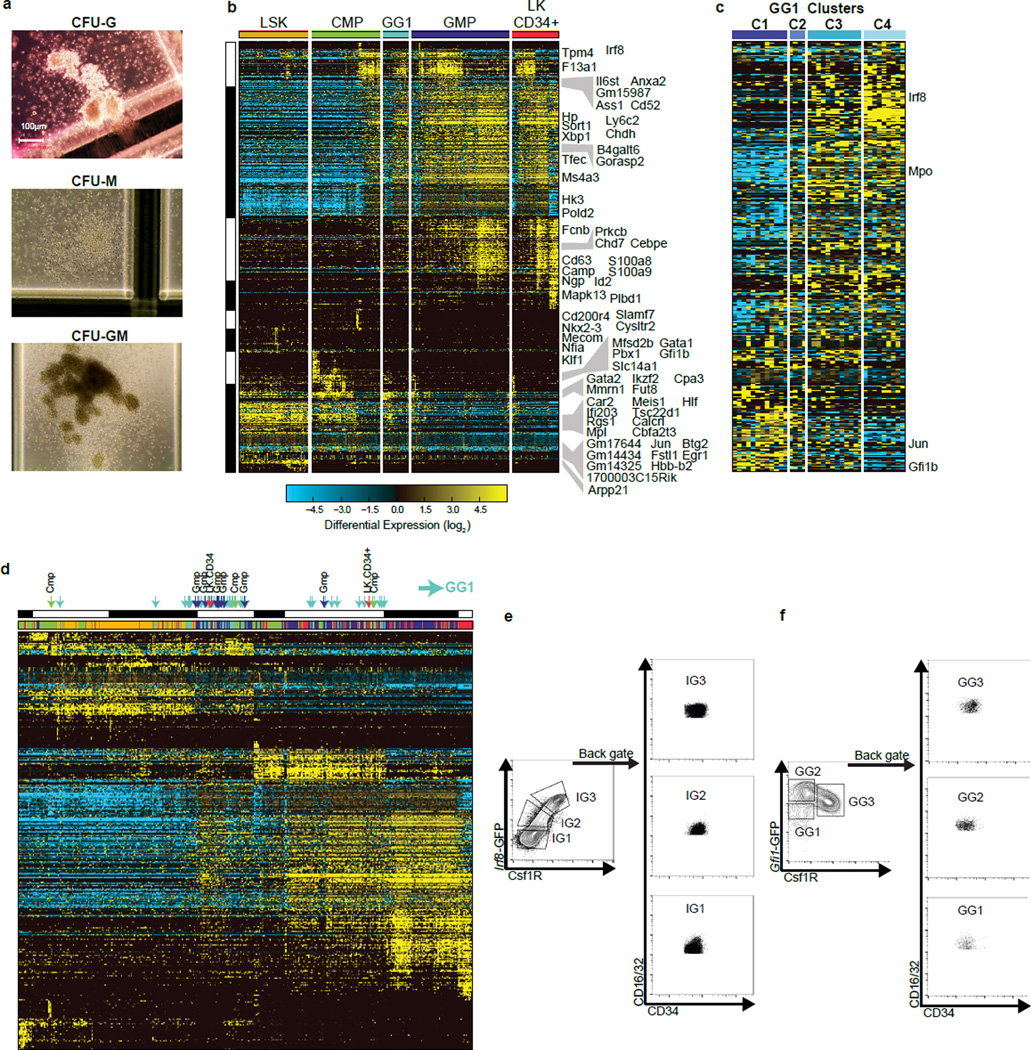
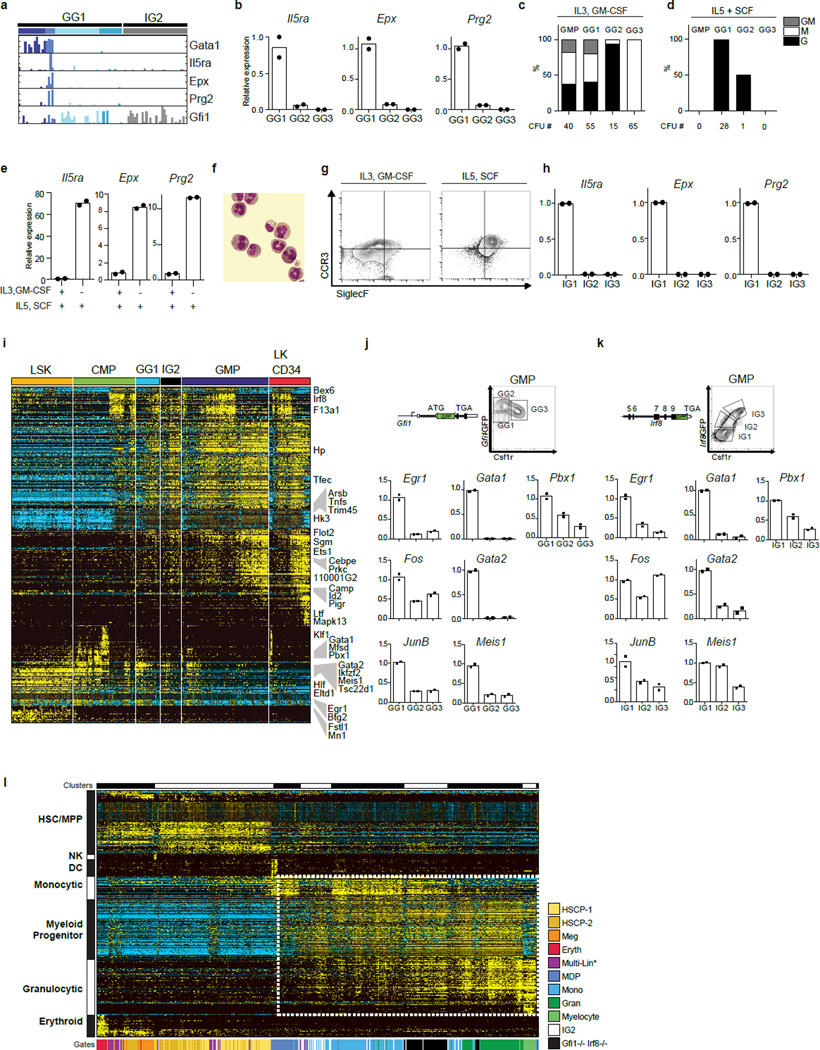
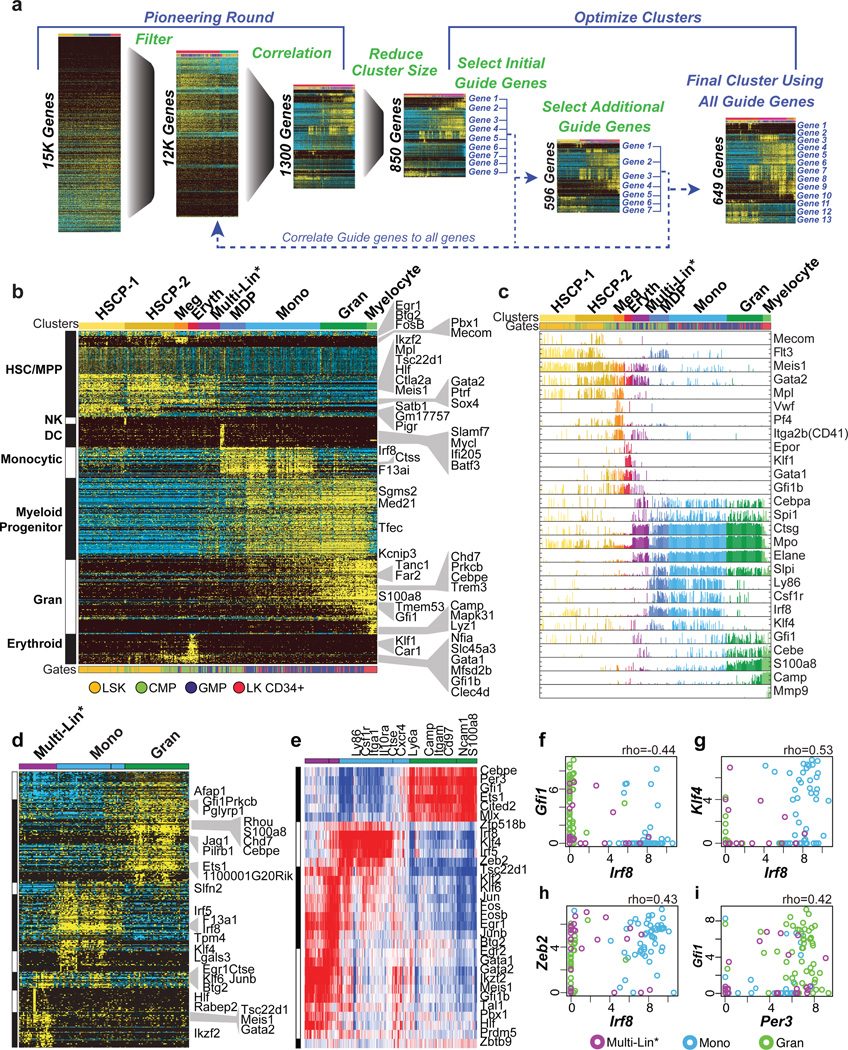
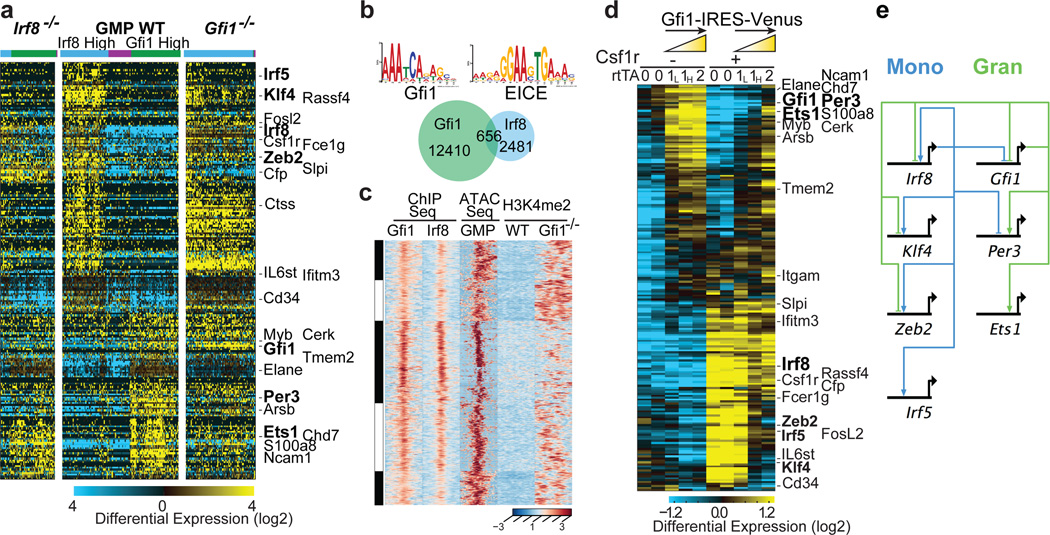
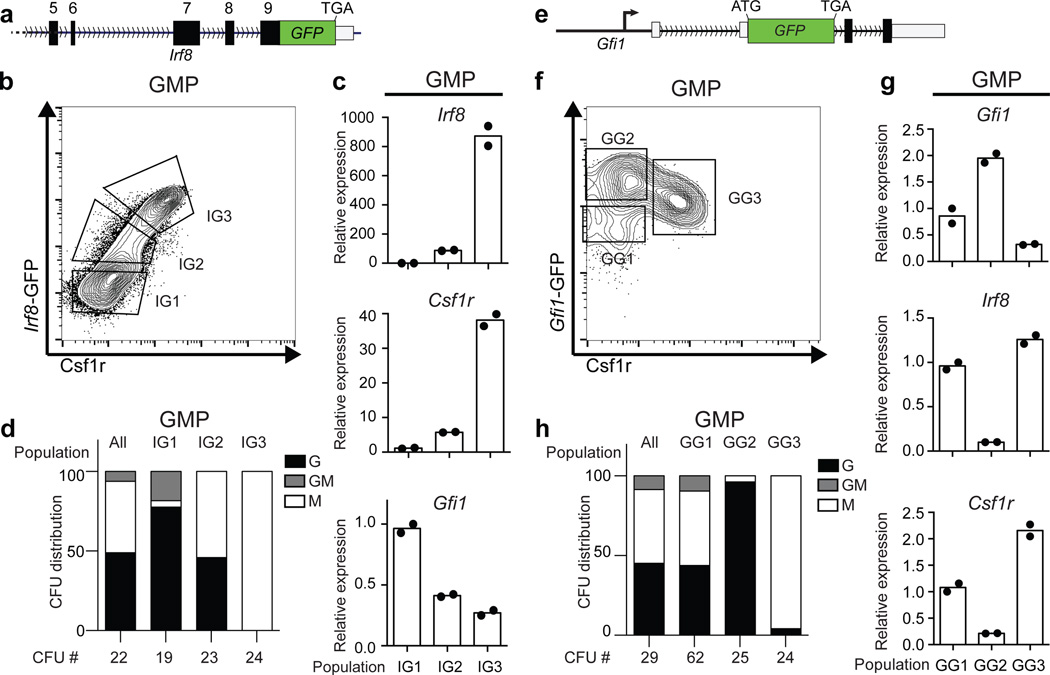
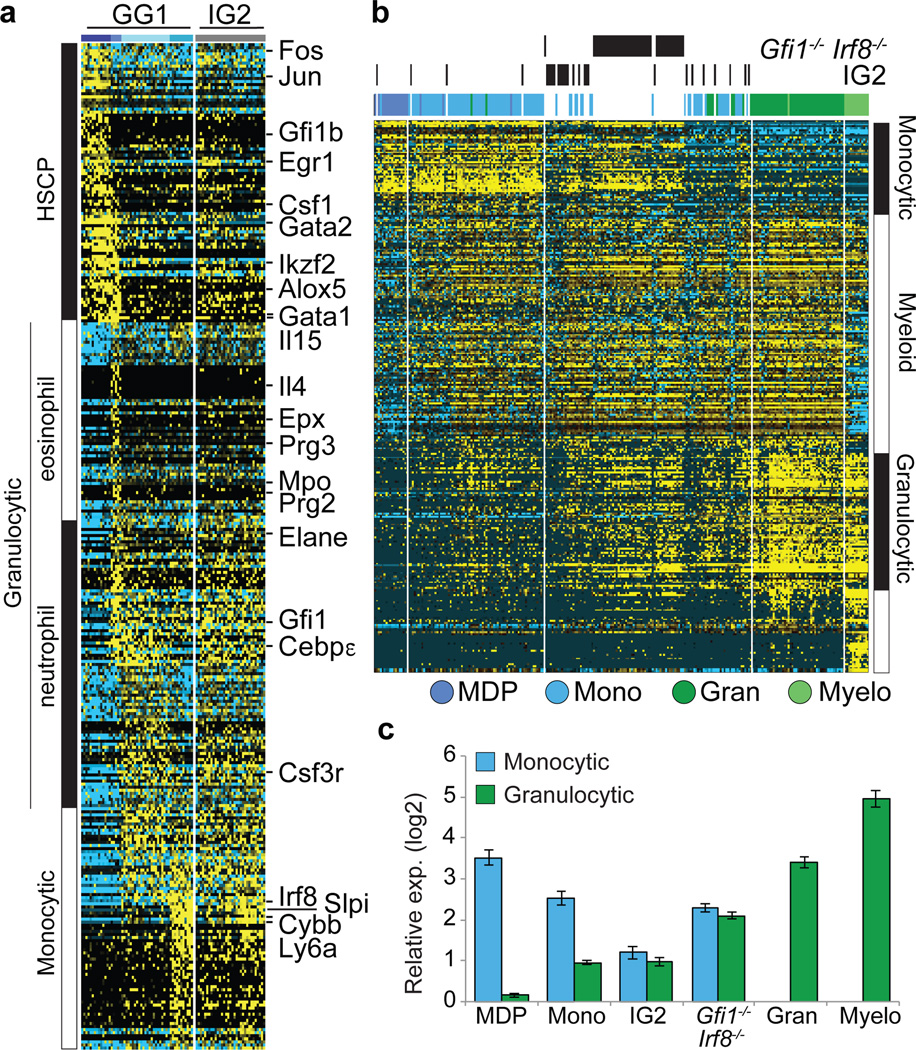
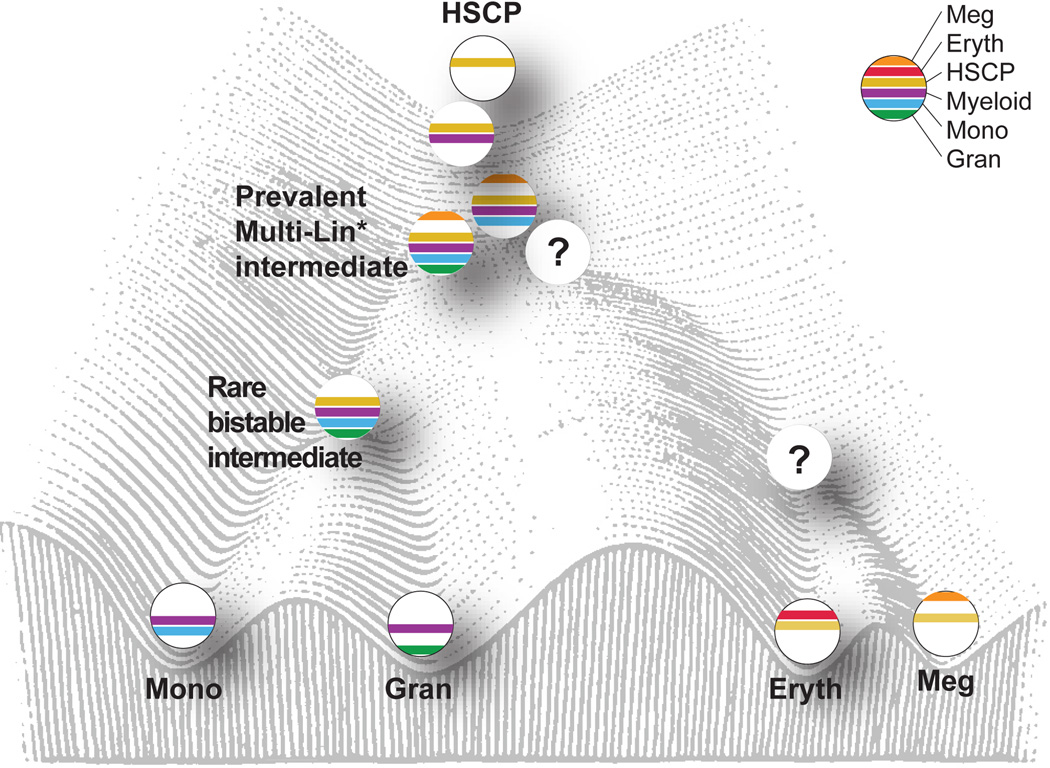
Delineating hierarchical cellular states, including rare intermediates and the networks of regulatory genes that orchestrate cell-type specification, are continuing challenges for developmental biology. Single-cell RNA sequencing is greatly accelerating such research, given its power to provide comprehensive descriptions of genomic states and their presumptive regulators. Haematopoietic multipotential progenitor cells, as well as bipotential intermediates, manifest mixed-lineage patterns of gene expression at a single-cell level. Such mixed-lineage states may reflect the molecular priming of different developmental potentials by co-expressed alternative-lineage determinants, namely transcription factors. Although a bistable gene regulatory network has been proposed to regulate the specification of either neutrophils or macrophages, the nature of the transition states manifested in vivo, and the underlying dynamics of the cell-fate determinants, have remained elusive. Here we use single-cell RNA sequencing coupled with a new analytic tool, iterative clustering and guide-gene selection, and clonogenic assays to delineate hierarchical genomic and regulatory states that culminate in neutrophil or macrophage specification in mice. We show that this analysis captured prevalent mixed-lineage intermediates that manifested concurrent expression of haematopoietic stem cell/progenitor and myeloid progenitor cell genes. It also revealed rare metastable intermediates that had collapsed the haematopoietic stem cell/progenitor gene expression programme, instead expressing low levels of the myeloid determinants, Irf8 and Gfi1 (refs 9, 10, 11, 12, 13). Genetic perturbations and chromatin immunoprecipitation followed by sequencing revealed Irf8 and Gfi1 as key components of counteracting myeloid-gene-regulatory networks. Combined loss of these two determinants 'trapped' the metastable intermediate. We propose that mixed-lineage states are obligatory during cell-fate specification, manifest differing frequencies because of their dynamic instability and are dictated by counteracting gene-regulatory networks.
Figures
References
-
- Grun D, et al. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature. 2015;525:251–255. - PubMed
-
- Paul F, et al. Transcriptional Heterogeneity and Lineage Commitment in Myeloid Progenitors. Cell. 2015;163:1663–1677. - PubMed
-
- Yan L, et al. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat Struct Mol Biol. 2013;20:1131–1139. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
