

Dual inhibition of complement and Toll-like receptors as a novel approach to treat inflammatory diseases-C3 or C5 emerge together with CD14 as promising targets
- PMID: 27581539
- PMCID: PMC5166441
- DOI: 10.1189/jlb.3VMR0316-132R
Dual inhibition of complement and Toll-like receptors as a novel approach to treat inflammatory diseases-C3 or C5 emerge together with CD14 as promising targets
Abstract
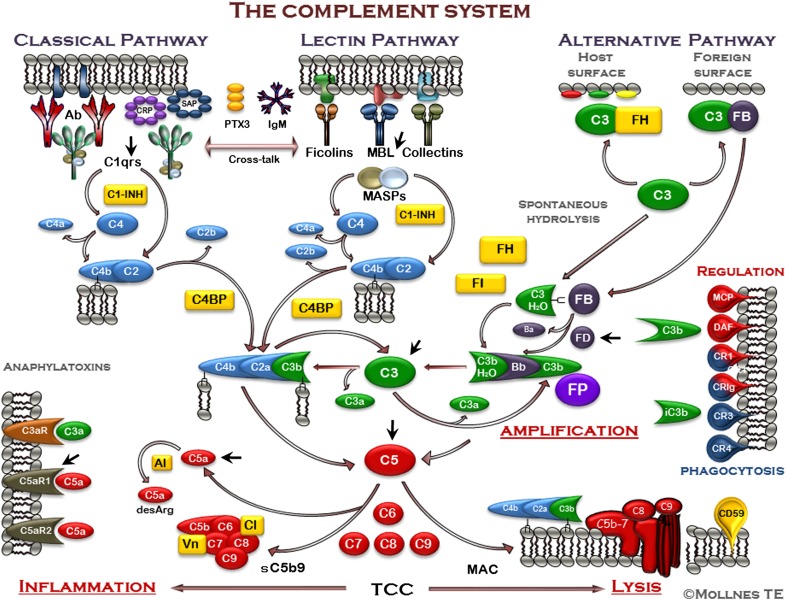
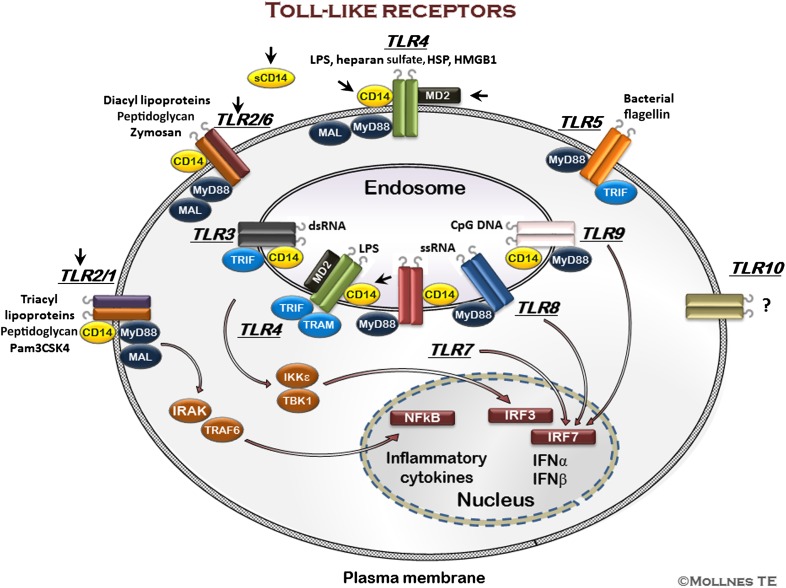
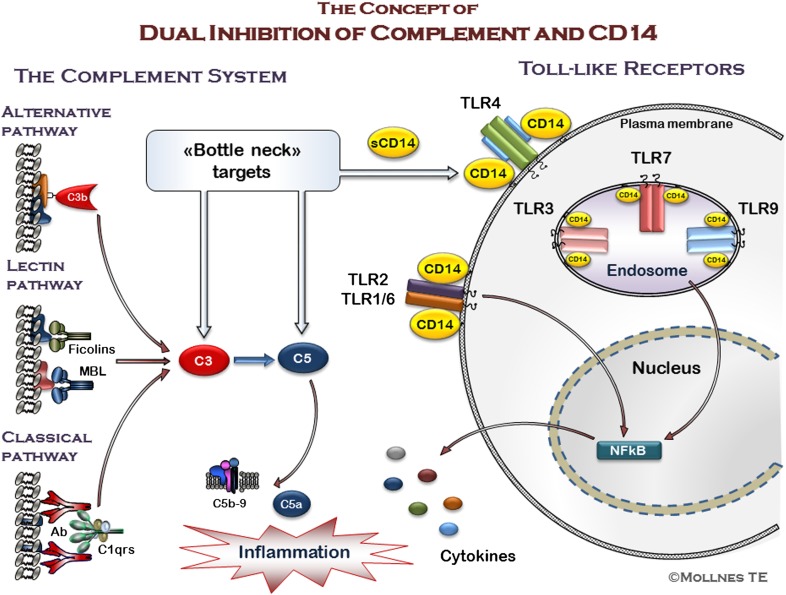
The host is protected by pattern recognition systems, including complement and TLRs, which are closely cross-talking. If improperly activated, these systems might induce tissue damage and disease. Inhibition of single downstream proinflammatory cytokines, such as TNF, IL-1β, and IL-6, have failed in clinical sepsis trials, which might not be unexpected, given the substantial amounts of mediators involved in the pathogenesis of this condition. Instead, we have put forward a hypothesis of inhibition at the recognition phase by "dual blockade" of bottleneck molecules of complement and TLRs. By acting upstream and broadly, the dual blockade could be beneficial in conditions with improper or uncontrolled innate immune activation threatening the host. Key bottleneck molecules in these systems that could be targets for inhibition are the central complement molecules C3 and C5 and the important CD14 molecule, which is a coreceptor for several TLRs, including TLR4 and TLR2. This review summarizes current knowledge of inhibition of complement and TLRs alone and in combination, in both sterile and nonsterile inflammatory processes, where activation of these systems is of crucial importance for tissue damage and disease. Thus, dual blockade might provide a general, broad-acting therapeutic regimen against a number of diseases where innate immunity is improperly activated.
Keywords: inflammation; innate immunity; therapy.
© The Author(s).
Figures
References
-
- Thaiss C. A., Levy M., Itav S., Elinav E. (2016) Integration of innate immune signaling. Trends Immunol. 37, 84–101. - PubMed
-
- Matzinger P. (1994) Tolerance, danger, and the extended family. Annu. Rev. Immunol. 12, 991–1045. - PubMed
-
- Sendide K., Reiner N. E., Lee J. S., Bourgoin S., Talal A., Hmama Z. (2005) Cross-talk between CD14 and complement receptor 3 promotes phagocytosis of mycobacteria: regulation by phosphatidylinositol 3-kinase and cytohesin-1. J. Immunol. 174, 4210–4219. - PubMed
-
- Köhl J. (2006) The role of complement in danger sensing and transmission. Immunol. Res. 34, 157–176. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
