

An analytical workflow for accurate variant discovery in highly divergent regions
- PMID: 27590916
- PMCID: PMC5010666
- DOI: 10.1186/s12864-016-3045-z
An analytical workflow for accurate variant discovery in highly divergent regions
Abstract
Background: Current variant discovery methods often start with the mapping of short reads to a reference genome; yet, their performance deteriorates in genomic regions where the reads are highly divergent from the reference sequence. This is particularly problematic for the human leukocyte antigen (HLA) region on chromosome 6p21.3. This region is associated with over 100 diseases, but variant calling is hindered by the extreme divergence across different haplotypes.
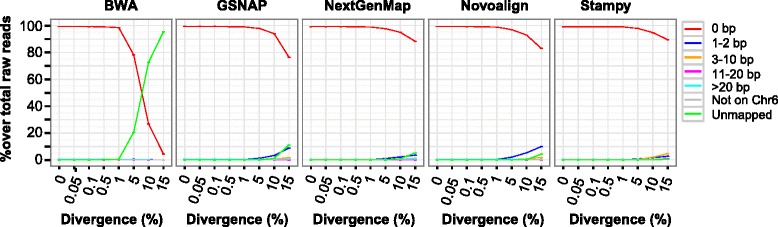
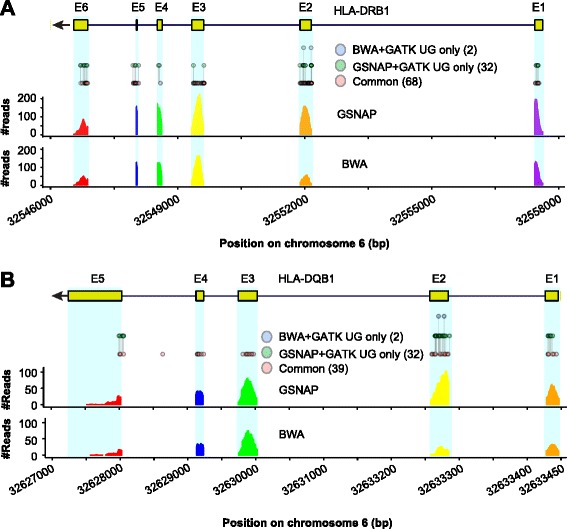
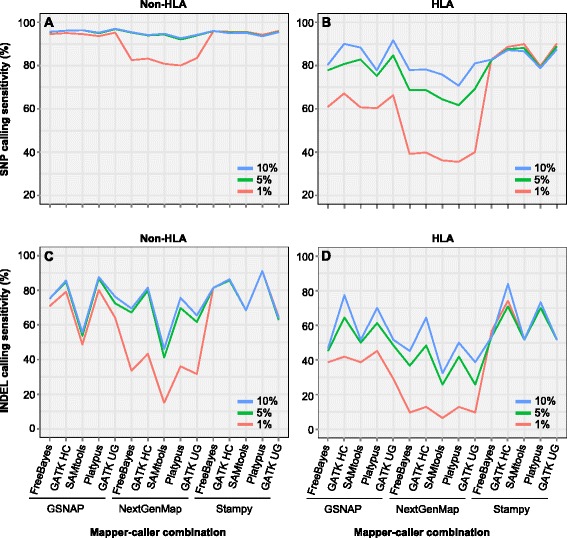
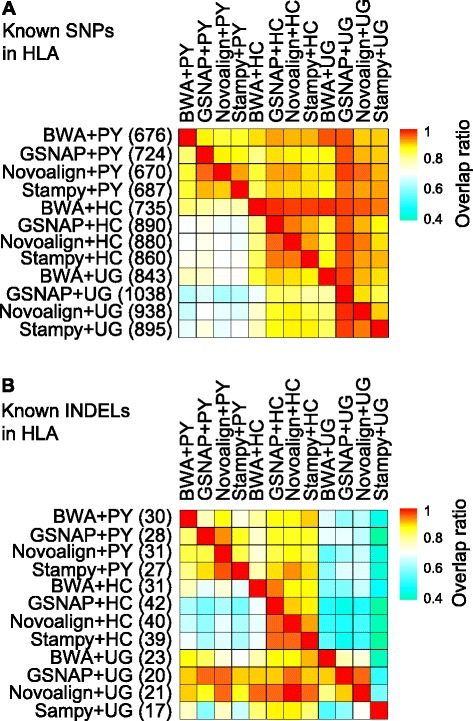
Results: We simulated reads from chromosome 6 exonic regions over a wide range of sequence divergence and coverage depth. We systematically assessed combinations between five mappers and five callers for their performance on simulated data and exome-seq data from NA12878, a well-studied individual in which multiple public call sets have been generated. Among those combinations, the number of known SNPs differed by about 5 % in the non-HLA regions of chromosome 6 but over 20 % in the HLA region. Notably, GSNAP mapping combined with GATK UnifiedGenotyper calling identified about 20 % more known SNPs than most existing methods without a noticeable loss of specificity, with 100 % sensitivity in three highly polymorphic HLA genes examined. Much larger differences were observed among these combinations in INDEL calling from both non-HLA and HLA regions. We obtained similar results with our internal exome-seq data from a cohort of chronic lymphocytic leukemia patients.
Conclusions: We have established a workflow enabling variant detection, with high sensitivity and specificity, over the full spectrum of divergence seen in the human genome. Comparing to public call sets from NA12878 has highlighted the overall superiority of GATK UnifiedGenotyper, followed by GATK HaplotypeCaller and SAMtools, in SNP calling, and of GATK HaplotypeCaller and Platypus in INDEL calling, particularly in regions of high sequence divergence such as the HLA region. GSNAP and Novoalign are the ideal mappers in combination with the above callers. We expect that the proposed workflow should be applicable to variant discovery in other highly divergent regions.
Keywords: Alignment algorithm; Chronic lymphocytic leukemia; Exome sequencing; Human leukocyte antigen; Variant calling.
Figures

Similar articles
-
Comparative analysis of de novo assemblers for variation discovery in personal genomes.Brief Bioinform. 2018 Sep 28;19(5):893-904. doi: 10.1093/bib/bbx037. Brief Bioinform. 2018. PMID: 28407084 Free PMC article.
-
Impact of post-alignment processing in variant discovery from whole exome data.BMC Bioinformatics. 2016 Oct 3;17(1):403. doi: 10.1186/s12859-016-1279-z. BMC Bioinformatics. 2016. PMID: 27716037 Free PMC article.
-
OVarFlow: a resource optimized GATK 4 based Open source Variant calling workFlow.BMC Bioinformatics. 2021 Aug 13;22(1):402. doi: 10.1186/s12859-021-04317-y. BMC Bioinformatics. 2021. PMID: 34388963 Free PMC article.
-
Toward better understanding of artifacts in variant calling from high-coverage samples.Bioinformatics. 2014 Oct 15;30(20):2843-51. doi: 10.1093/bioinformatics/btu356. Epub 2014 Jun 27. Bioinformatics. 2014. PMID: 24974202 Free PMC article. Review.
-
A beginners guide to SNP calling from high-throughput DNA-sequencing data.Hum Genet. 2012 Oct;131(10):1541-54. doi: 10.1007/s00439-012-1213-z. Epub 2012 Aug 11. Hum Genet. 2012. PMID: 22886560 Review.
Cited by
-
Comparative analysis of de novo assemblers for variation discovery in personal genomes.Brief Bioinform. 2018 Sep 28;19(5):893-904. doi: 10.1093/bib/bbx037. Brief Bioinform. 2018. PMID: 28407084 Free PMC article.
-
A comparison of genotyping-by-sequencing analysis methods on low-coverage crop datasets shows advantages of a new workflow, GB-eaSy.BMC Bioinformatics. 2017 Dec 28;18(1):586. doi: 10.1186/s12859-017-2000-6. BMC Bioinformatics. 2017. PMID: 29281959 Free PMC article.
-
UNISOM: Unified Somatic Calling and Machine Learning-based Classification Enhance the Discovery of CHIP.Genomics Proteomics Bioinformatics. 2025 May 30;23(2):qzaf040. doi: 10.1093/gpbjnl/qzaf040. Genomics Proteomics Bioinformatics. 2025. PMID: 40300108 Free PMC article.
-
Genomic diversity affects the accuracy of bacterial single-nucleotide polymorphism-calling pipelines.Gigascience. 2020 Feb 1;9(2):giaa007. doi: 10.1093/gigascience/giaa007. Gigascience. 2020. PMID: 32025702 Free PMC article.
-
Performance evaluation of pipelines for mapping, variant calling and interval padding, for the analysis of NGS germline panels.BMC Bioinformatics. 2021 Apr 28;22(1):218. doi: 10.1186/s12859-021-04144-1. BMC Bioinformatics. 2021. PMID: 33910496 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
