

Development of a Comprehensive Genotype-to-Fitness Map of Adaptation-Driving Mutations in Yeast
- PMID: 27594428
- PMCID: PMC5070919
- DOI: 10.1016/j.cell.2016.08.002
Development of a Comprehensive Genotype-to-Fitness Map of Adaptation-Driving Mutations in Yeast
Abstract
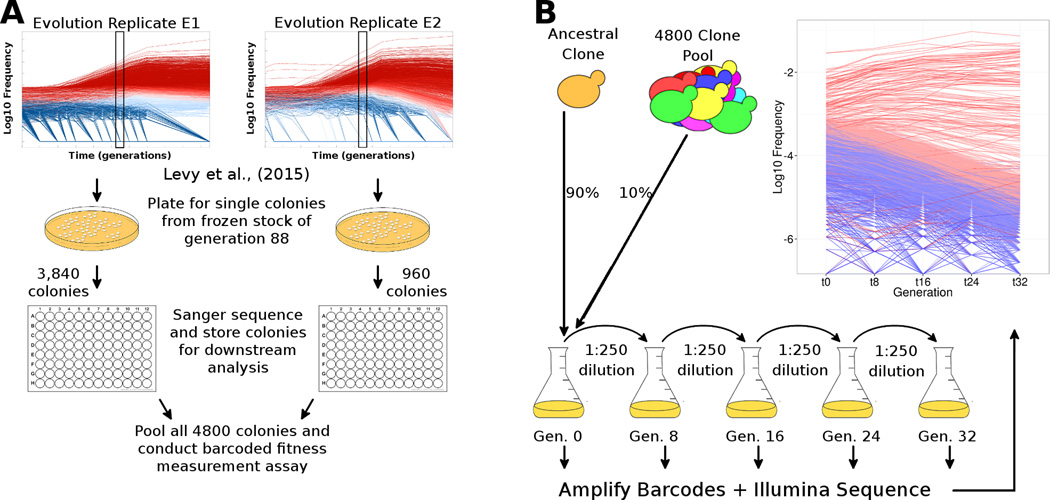
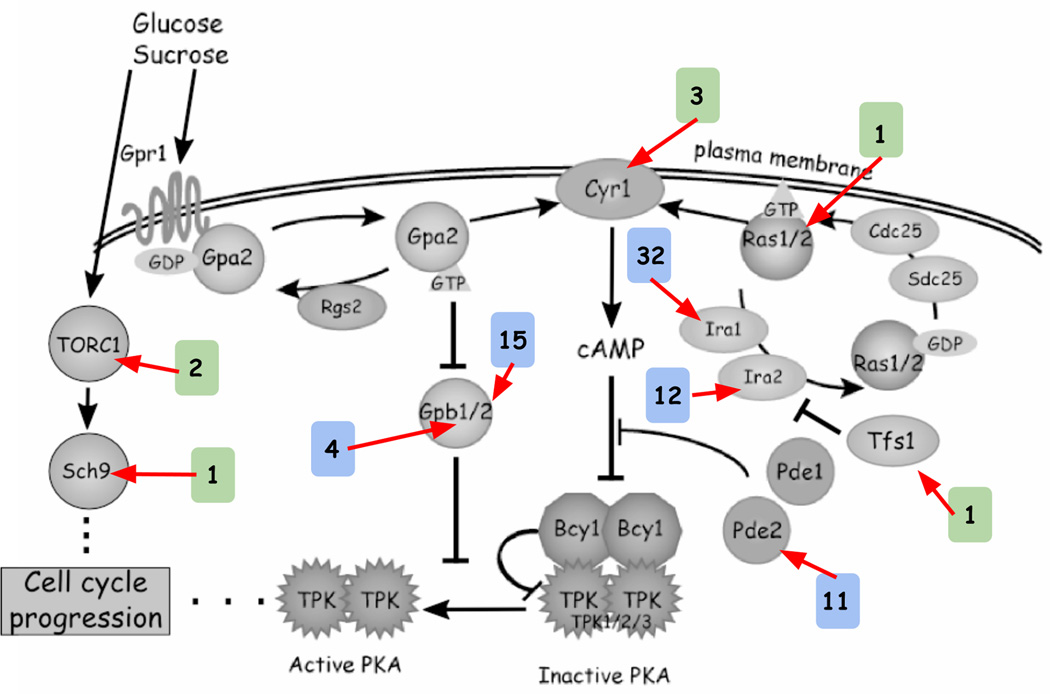
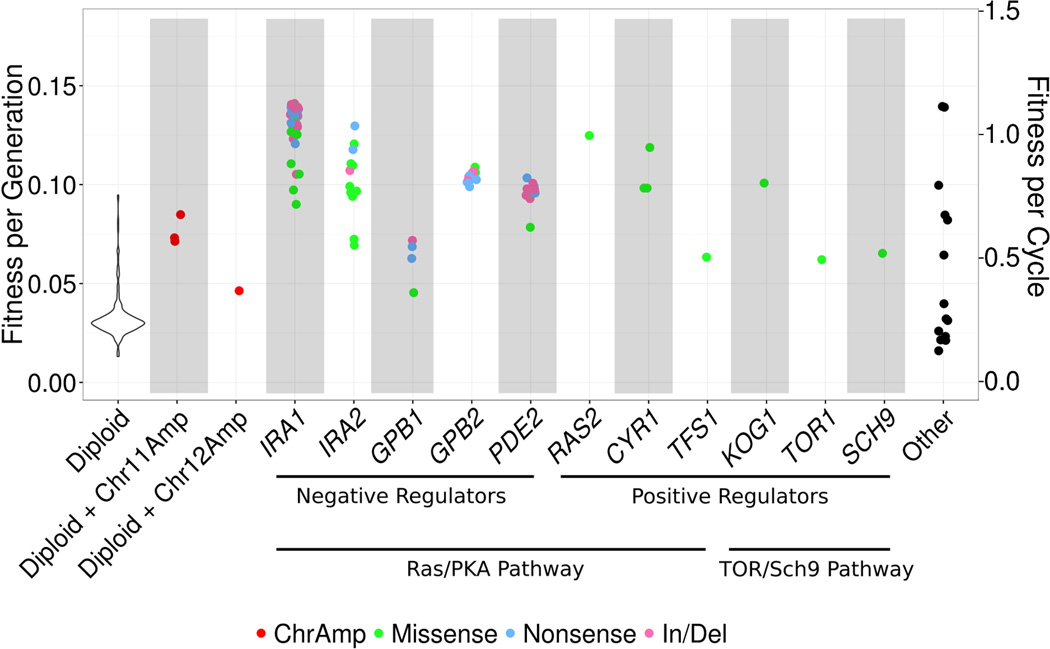
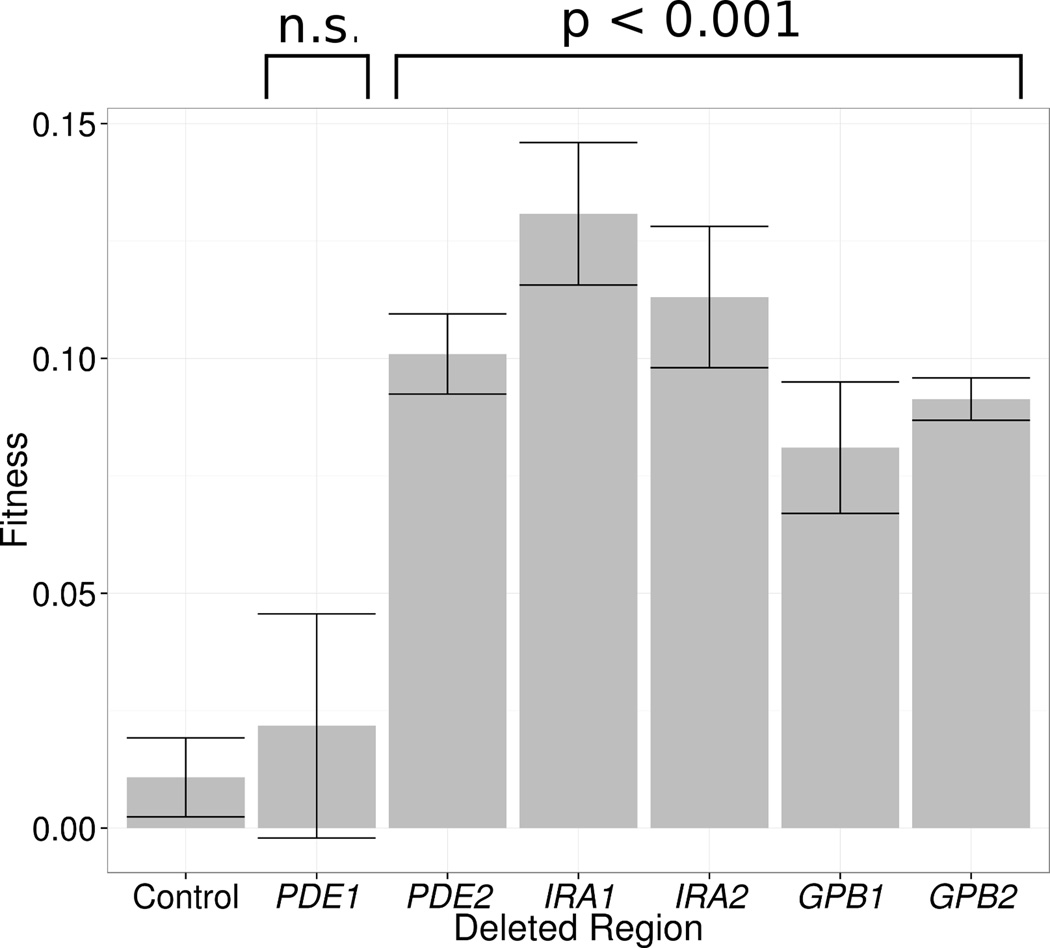
Adaptive evolution plays a large role in generating the phenotypic diversity observed in nature, yet current methods are impractical for characterizing the molecular basis and fitness effects of large numbers of individual adaptive mutations. Here, we used a DNA barcoding approach to generate the genotype-to-fitness map for adaptation-driving mutations from a Saccharomyces cerevisiae population experimentally evolved by serial transfer under limiting glucose. We isolated and measured the fitness of thousands of independent adaptive clones and sequenced the genomes of hundreds of clones. We found only two major classes of adaptive mutations: self-diploidization and mutations in the nutrient-responsive Ras/PKA and TOR/Sch9 pathways. Our large sample size and precision of measurement allowed us to determine that there are significant differences in fitness between mutations in different genes, between different paralogs, and even between different classes of mutations within the same gene.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures

References
-
- Barrick JE, Yu DS, Yoon SH, Jeong H, Oh TK, Schneider D, Lenski RE, Kim JF. Genome evolution and adaptation in a long-term experiment with Escherichia coli . Nature. 2009;461:1243–1247. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
