

Viral deep sequencing needs an adaptive approach: IRMA, the iterative refinement meta-assembler
- PMID: 27595578
- PMCID: PMC5011931
- DOI: 10.1186/s12864-016-3030-6
Viral deep sequencing needs an adaptive approach: IRMA, the iterative refinement meta-assembler
Erratum in
-
Erratum to: Viral deep sequencing needs an adaptive approach: IRMA, the iterative refinement meta-assembler.BMC Genomics. 2016 Oct 13;17(1):801. doi: 10.1186/s12864-016-3138-8. BMC Genomics. 2016. PMID: 27737640 Free PMC article. No abstract available.
Abstract
Background: Deep sequencing makes it possible to observe low-frequency viral variants and sub-populations with greater accuracy and sensitivity than ever before. Existing platforms can be used to multiplex a large number of samples; however, analysis of the resulting data is complex and involves separating barcoded samples and various read manipulation processes ending in final assembly. Many assembly tools were designed with larger genomes and higher fidelity polymerases in mind and do not perform well with reads derived from highly variable viral genomes. Reference-based assemblers may leave gaps in viral assemblies while de novo assemblers may struggle to assemble unique genomes.
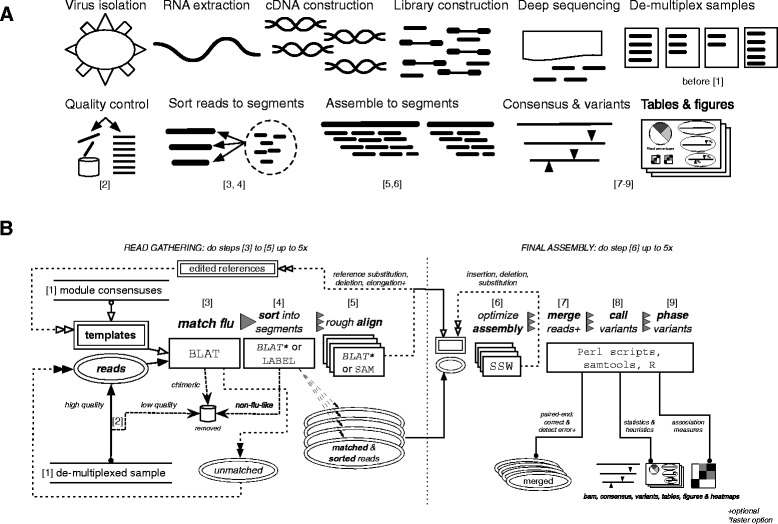
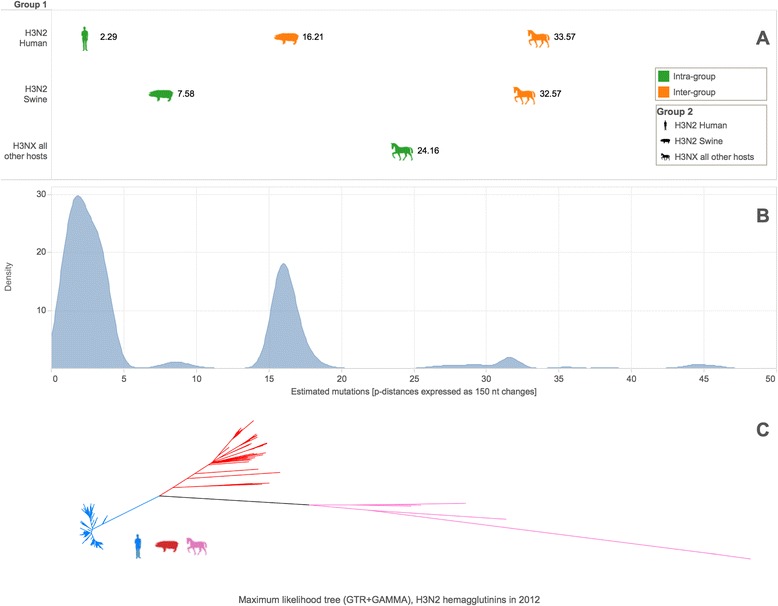
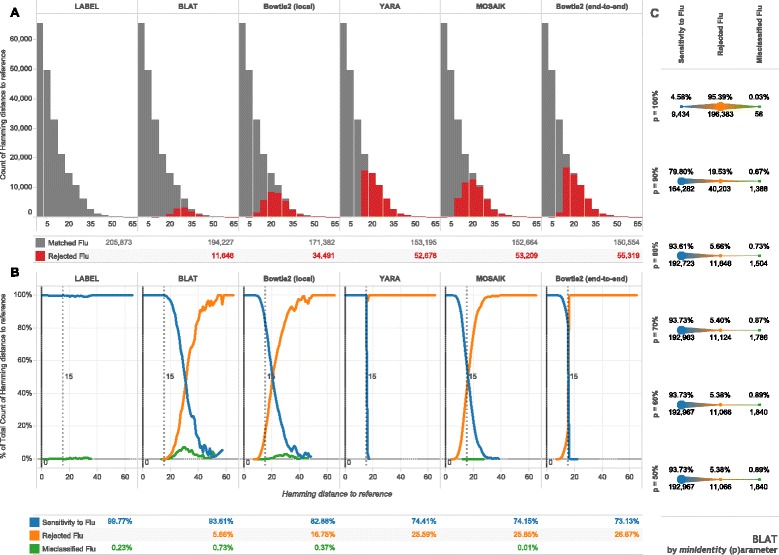
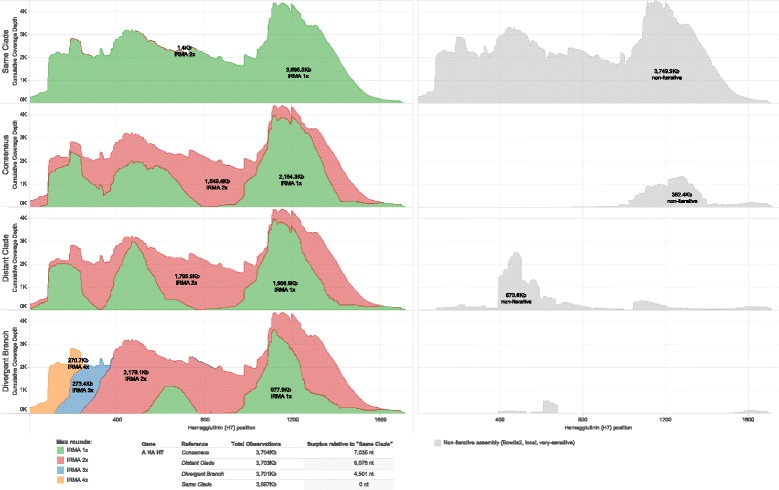
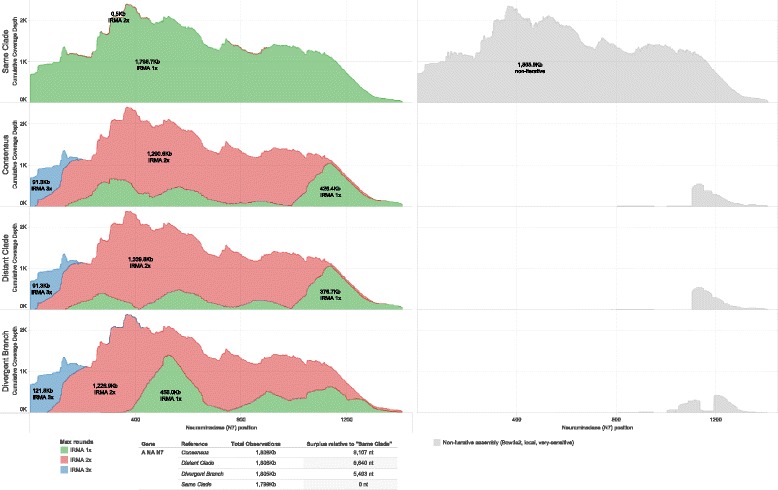
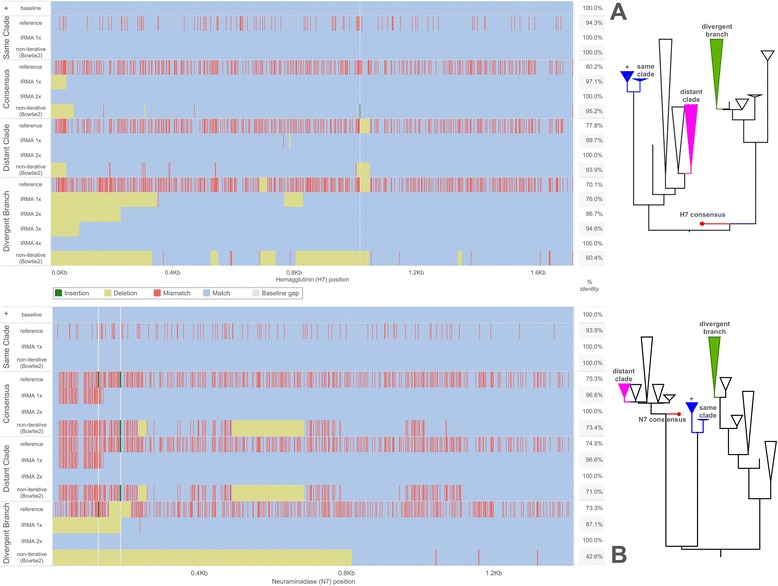
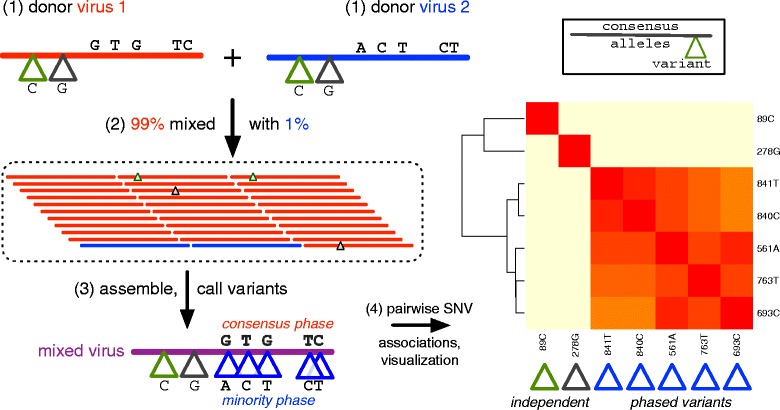
Results: The IRMA (iterative refinement meta-assembler) pipeline solves the problem of viral variation by the iterative optimization of read gathering and assembly. As with all reference-based assembly, reads are included in assembly when they match consensus template sets; however, IRMA provides for on-the-fly reference editing, correction, and optional elongation without the need for additional reference selection. This increases both read depth and breadth. IRMA also focuses on quality control, error correction, indel reporting, variant calling and variant phasing. In fact, IRMA's ability to detect and phase minor variants is one of its most distinguishing features. We have built modules for influenza and ebolavirus. We demonstrate usage and provide calibration data from mixture experiments. Methods for variant calling, phasing, and error estimation/correction have been redesigned to meet the needs of viral genomic sequencing.
Conclusion: IRMA provides a robust next-generation sequencing assembly solution that is adapted to the needs and characteristics of viral genomes. The software solves issues related to the genetic diversity of viruses while providing customized variant calling, phasing, and quality control. IRMA is freely available for non-commercial use on Linux and Mac OS X and has been parallelized for high-throughput computing.
Keywords: Deep sequencing; Ebola; High throughput; Influenza; NGS; Public health; Surveillance.
Figures
References
-
- Reed C, Chaves SS, Daily Kirley P, Emerson R, Aragon D, Hancock EB, Butler L, Baumbach J, Hollick G, Bennett NM, et al. Estimating influenza disease burden from population-based surveillance data in the United States. PLoS One. 2015;10(3):e0118369. doi: 10.1371/journal.pone.0118369. - DOI - PMC - PubMed
-
- FluNet: total influenza A and B specimens detected. [http://www.who.int/influenza/gisrs_laboratory/flunet]. Accessed 7 Nov 2015.
-
- Westgeest KB, Russell CA, Lin X, Spronken MI, Bestebroer TM, Bahl J, van Beek R, Skepner E, Halpin RA, de Jong JC, et al. Genomewide analysis of reassortment and evolution of human influenza A(H3N2) viruses circulating between 1968 and 2011. J Virol. 2014;88(5):2844–57. doi: 10.1128/JVI.02163-13. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
