

GM-CSF primes cardiac inflammation in a mouse model of Kawasaki disease
- PMID: 27595596
- PMCID: PMC5030799
- DOI: 10.1084/jem.20151853
GM-CSF primes cardiac inflammation in a mouse model of Kawasaki disease
Abstract
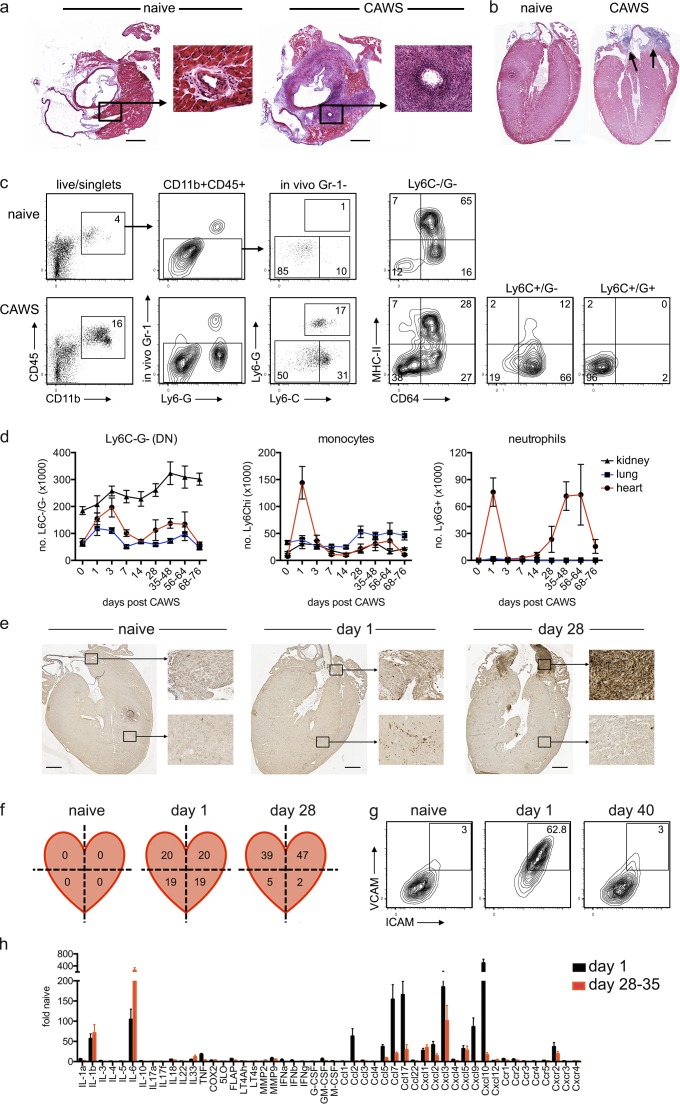
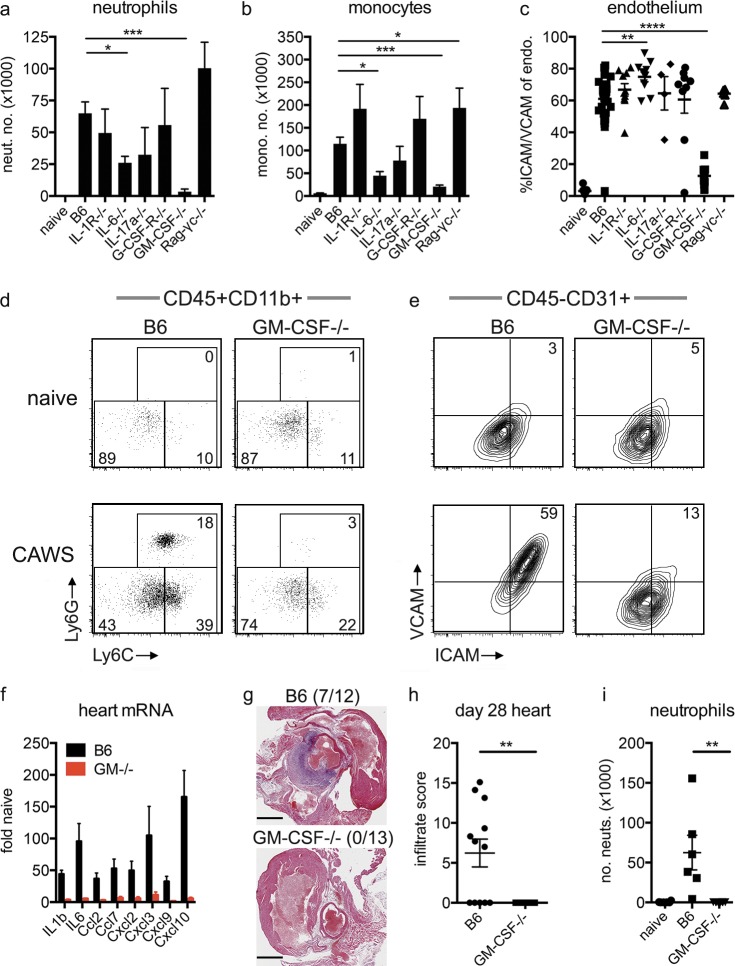
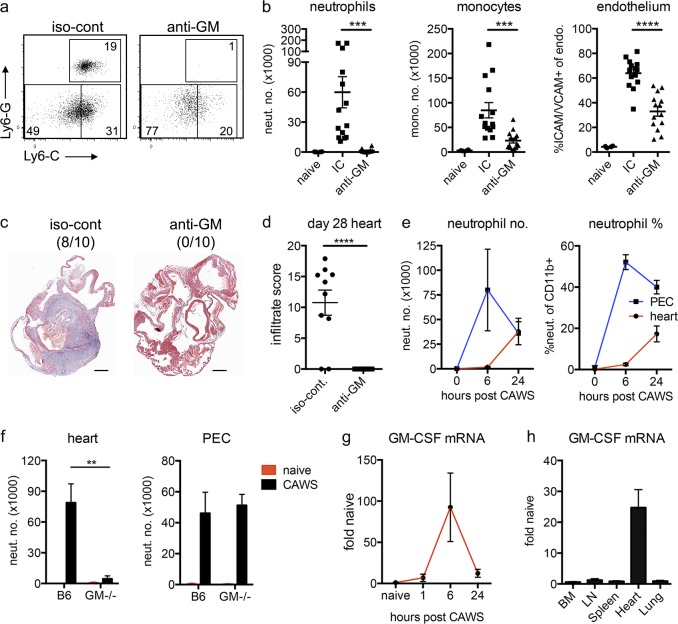
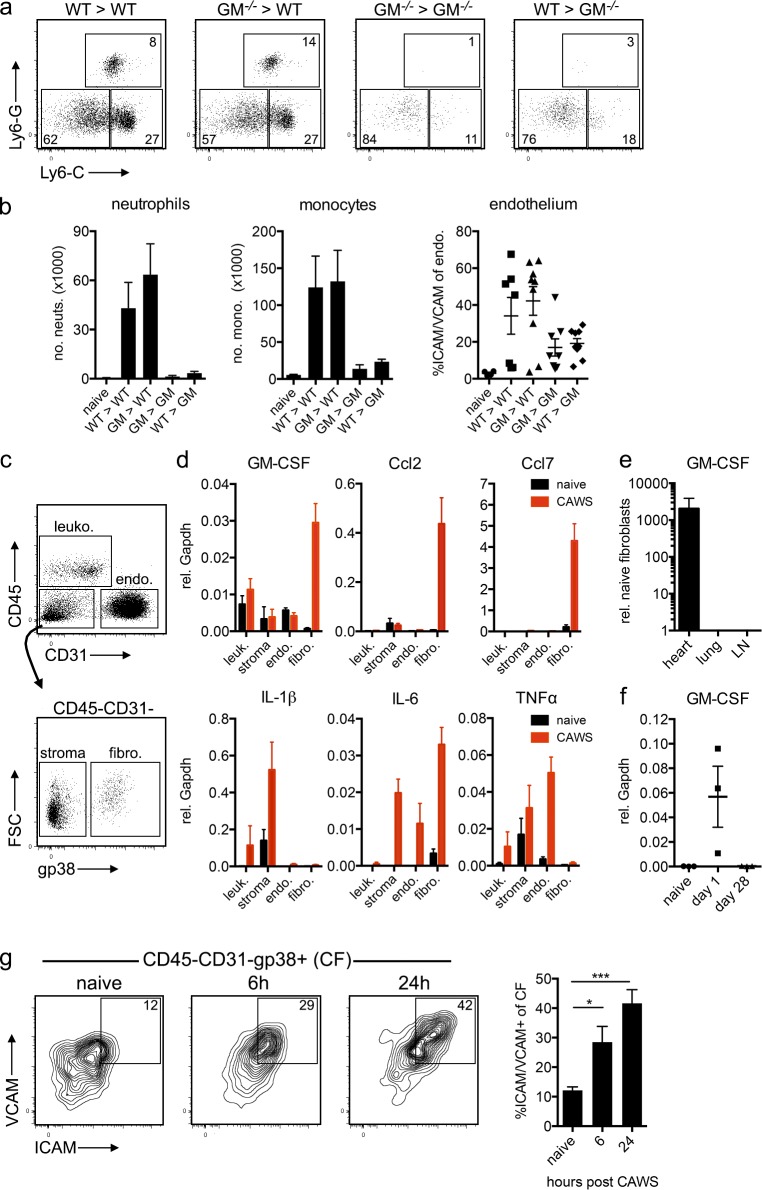
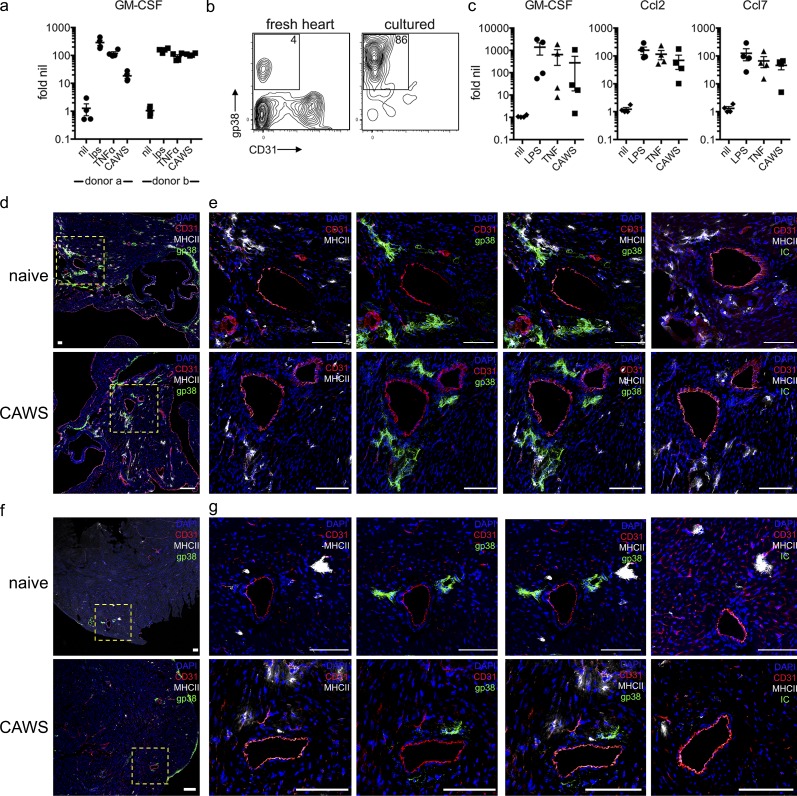
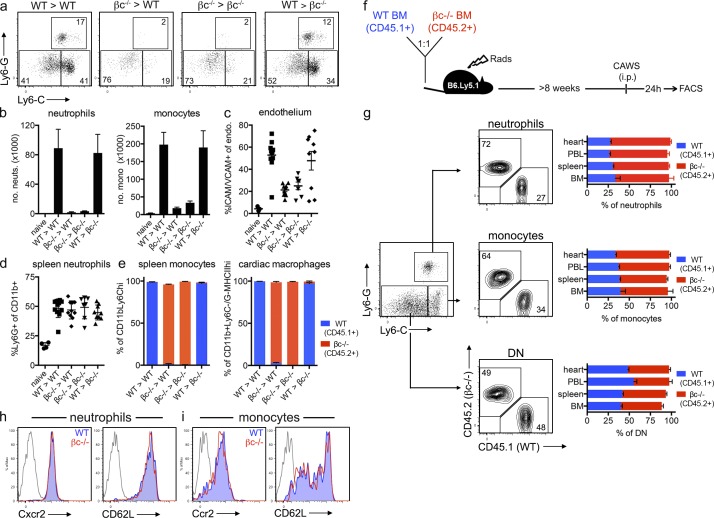
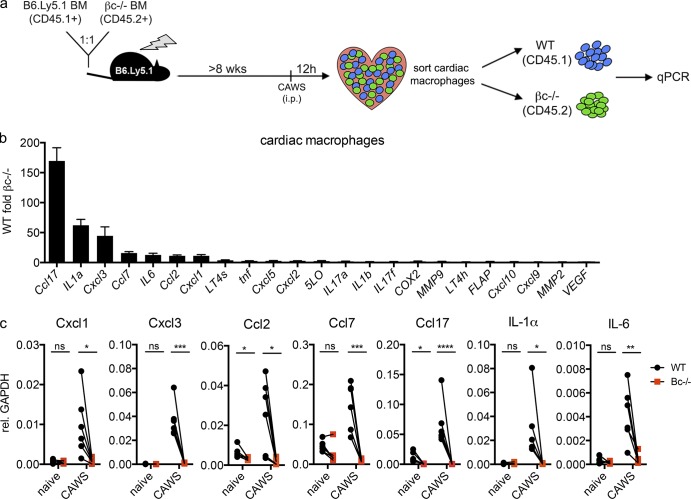
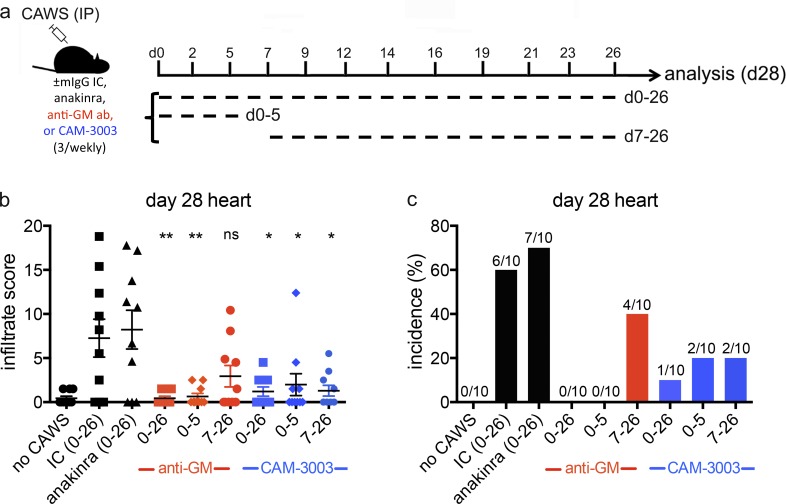
Kawasaki disease (KD) is the leading cause of pediatric heart disease in developed countries. KD patients develop cardiac inflammation, characterized by an early infiltrate of neutrophils and monocytes that precipitates coronary arteritis. Although the early inflammatory processes are linked to cardiac pathology, the factors that regulate cardiac inflammation and immune cell recruitment to the heart remain obscure. In this study, using a mouse model of KD (induced by a cell wall Candida albicans water-soluble fraction [CAWS]), we identify an essential role for granulocyte/macrophage colony-stimulating factor (GM-CSF) in orchestrating these events. GM-CSF is rapidly produced by cardiac fibroblasts after CAWS challenge, precipitating cardiac inflammation. Mechanistically, GM-CSF acts upon the local macrophage compartment, driving the expression of inflammatory cytokines and chemokines, whereas therapeutically, GM-CSF blockade markedly reduces cardiac disease. Our findings describe a novel role for GM-CSF as an essential initiating cytokine in cardiac inflammation and implicate GM-CSF as a potential target for therapeutic intervention in KD.
© 2016 Stock et al.
Figures
Similar articles
-
Adeno-associated Virus Vector-mediated Interleukin-10 Induction Prevents Vascular Inflammation in a Murine Model of Kawasaki Disease.Sci Rep. 2018 May 15;8(1):7601. doi: 10.1038/s41598-018-25856-0. Sci Rep. 2018. PMID: 29765083 Free PMC article.
-
Macrophage-colony stimulating factor is required for the production of neutrophil-promoting activity by mouse embryo fibroblasts deficient in G-CSF and GM-CSF.J Leukoc Biol. 2007 Oct;82(4):915-25. doi: 10.1189/jlb.0107023. Epub 2007 Jul 25. J Leukoc Biol. 2007. PMID: 17652450
-
Characterization of a murine model with arteritis induced by Nod1 ligand, FK565: A comparative study with a CAWS-induced model.Mod Rheumatol. 2017 Nov;27(6):1024-1030. doi: 10.1080/14397595.2017.1287150. Epub 2017 Feb 21. Mod Rheumatol. 2017. PMID: 28150515
-
Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: what we do and don't know.Cell Res. 2006 Feb;16(2):126-33. doi: 10.1038/sj.cr.7310017. Cell Res. 2006. PMID: 16474424 Review.
-
Extra-pulmonary aspects of acquired pulmonary alveolar proteinosis as predicted by granulocyte-macrophage colony-stimulating factor-deficient mice.Respirology. 2006 Jan;11 Suppl:S16-22. doi: 10.1111/j.1440-1843.2006.00801.x. Respirology. 2006. PMID: 16423263 Review.
Cited by
-
GM-CSF: A Promising Target in Inflammation and Autoimmunity.Immunotargets Ther. 2020 Oct 29;9:225-240. doi: 10.2147/ITT.S262566. eCollection 2020. Immunotargets Ther. 2020. PMID: 33150139 Free PMC article. Review.
-
CDCP1 on Dendritic Cells Contributes to the Development of a Model of Kawasaki Disease.J Immunol. 2021 Jun 15;206(12):2819-2827. doi: 10.4049/jimmunol.2001406. Epub 2021 Jun 7. J Immunol. 2021. PMID: 34099547 Free PMC article.
-
The Immune and Inflammatory Basis of Acquired Pediatric Cardiac Disease.Front Cardiovasc Med. 2021 Jul 27;8:701224. doi: 10.3389/fcvm.2021.701224. eCollection 2021. Front Cardiovasc Med. 2021. PMID: 34386532 Free PMC article. Review.
-
Role of the β Common (βc) Family of Cytokines in Health and Disease.Cold Spring Harb Perspect Biol. 2018 Jun 1;10(6):a028514. doi: 10.1101/cshperspect.a028514. Cold Spring Harb Perspect Biol. 2018. PMID: 28716883 Free PMC article. Review.
-
Clonal expansion and activation of tissue-resident memory-like Th17 cells expressing GM-CSF in the lungs of severe COVID-19 patients.Sci Immunol. 2021 Feb 23;6(56):eabf6692. doi: 10.1126/sciimmunol.abf6692. Sci Immunol. 2021. PMID: 33622974 Free PMC article.
References
-
- Biezeveld M.H., van Mierlo G., Lutter R., Kuipers I.M., Dekker T., Hack C.E., Newburger J.W., and Kuijpers T.W.. 2005. Sustained activation of neutrophils in the course of Kawasaki disease: an association with matrix metalloproteinases. Clin. Exp. Immunol. 141:183–188. 10.1111/j.1365-2249.2005.02829.x - DOI - PMC - PubMed
-
- Campbell I.K., van Nieuwenhuijze A., Segura E., O’Donnell K., Coghill E., Hommel M., Gerondakis S., Villadangos J.A., and Wicks I.P.. 2011. Differentiation of inflammatory dendritic cells is mediated by NF-κB1–dependent GM-CSF production in CD4 T cells. J. Immunol. 186:5468–5477. 10.4049/jimmunol.1002923 - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
